石油管道輸送用高效減阻劑超高分子量聚1-辛烯的合成及其結構性能
2023-03-15 03:32:10李春漫
石油化工 2023年2期
關鍵詞:催化劑
李春漫
(國家管網集團科學技術研究總院分公司,河北 廊坊 065000)
管道運輸具有成本低等優點,在油料運輸中發揮著重要作用。根據《2020年中國油氣管道建設和新進展》報道[1],我國油料管道運輸總里程數已近6×105km,管道運輸關系著我國能源安全問題。分子量高達數百萬的1-辛烯等長鏈α-烯烴聚合物能長時間保持優良的減阻效果[2],可用作石油管道輸送用減阻劑。開發超高分子量聚1-辛烯的核心技術是催化劑的設計與優化,而烯烴配位聚合催化劑有茂金屬催化劑[3-9]、高效Ziegler-Natta(Z-N)催化劑[10-14]和非茂金屬催化劑[15-18]。其中,茂金屬催化劑難以獲得高分子量α-烯烴聚合物;高效Z-N催化劑催化長鏈α-烯烴聚合的報道較多[10],但Z-N催化劑活性組分含量低,聚合效率低;非茂金屬催化劑在聚合時易發生β-H鏈轉移,也難以得到高分子量聚烯烴,故主要用于乙烯與長鏈α-烯烴共聚[19-23]。因此,需要在催化劑結構設計方面突破慣性思維,合成不易發生β-H鏈轉移的催化劑,以獲得高分子量聚烯烴。受烯烴配位聚合催化劑活性位點調控理論的啟發:在催化體系的結構/組成中引入強吸電子基團,調控過渡金屬原子周圍的電子效應與配位環境,可增強活性中心穩定性;增強活性中心與烯烴單體配位、插入及鏈增長的能力,可提高催化劑活性;提高烯烴聚合時β-H鏈轉移活化能,使β-H消除反應較難發生,可獲得高分子量聚烯烴。
本工作以苯酚及其衍生物為原料合成了5種催化劑,采用低溫常壓液相本體法,將催化劑催化1-辛烯聚合得到超高分子量聚1-辛烯減阻劑,利用NMR,XPS,DSC,FTIR,XRD等方法對催化劑結構進行了表征,考察了聚合條件對聚合反應的影響,并分析了聚1-辛烯的結構及減阻性能。
1 實驗部分
1.1 主要試劑
TiCl4(純度99%(w))、正丁基鋰(2.4 mol/L己烷溶液):上海阿拉丁生化科技股份有限公司;2,4,6-三甲基苯酚、對甲基苯酚、對氟苯酚、2,4,6-三氟苯酚:分析純,西格瑪奧德里奇(上海)貿易有限公司;二甲基亞砜(DMSO):分析純,Cambridge Isotope Laboratories,Inc.;氘代甲苯:分析純,北京伊諾凱科技有限公司;正己烷、甲苯、1-辛烯:分析純,上海阿拉丁生化科技股份有限公司,金屬鈉回流48 h后待用;甲基鋁氧烷(MAO):10%(w)甲苯溶液,國藥集團化學試劑有限公司。
1.2 表征方法
NMR分析采用Bruker公司AV400型核磁共振波譜儀,溫度40 ℃,溶劑為氘代DMSO。采用THERMO VG公司ESCALAB 250型X射線光電子能譜儀測定催化劑各元素含量。DSC分析采用TA公司DTG-Q50型差示掃描量熱儀,溫度范圍-80~200 ℃,升溫速率10 ℃/min,降溫速率20 ℃/min,取第2次升溫曲線。WAXD分析采用Bruker公司D2Phaser型X射線衍射儀,CuKα射線,波長1.540 5×10-10m,掃描范圍5°~50°。采用Julabo公司VISCO 370型黏度測試儀測定特性黏度,溶劑為甲苯,溫度30 ℃,根據特性黏度計算產物黏均分子量(Mη)。
按 SY/T 6578—2016[24]規定的方法,采用自行設計的室內測試環道開展減阻性能測試。
1.3 催化劑合成與表征
以苯酚及其衍生物、TiCl4為主要原料,合成催化劑Cat.1~Cat.5,催化劑合成路線見式(1),合成方法為:取1個250 mL Schlenk反應瓶,氮氣置換4~6次,用熱風槍烘烤15 min;冷卻至室溫后在氮氣氛圍下加入2,4,6-三甲基苯酚1.37 g(10.08 mmol)和甲苯40 mL;將反應瓶放入-10℃的冰鹽浴中,滴加正丁基鋰4.2 mL(2.4 mol/L己烷溶液),反應1 h后升至室溫反應3 h,得到微黃色對甲基苯酚鋰化合物甲苯溶液。再將反應瓶放入冰鹽浴(-10 ℃)中,用注射器滴加1.1 mL TiCl4,反應1 h后再加熱至50 ℃反應5 h。反應瓶放置冰箱中靜置結晶。室溫過濾,真空抽除殘余溶劑,氮氣保護,得到亮黃色固體產物C9H11OTiCl3(Cat.1)2.79 g,收率96.5%。
Cat.1(C9H11OTiCl3,分子量289.3)的XPS元素分析為:C fund 38.1 cal.37.3,Ti fund 15.8 cal.16.5,Cl fund 35.7 cal.36.8;1H NMR (400 MHz,DMSO)化學位移(δ)為:2.18(CH3,s,9H),6.79(CH,s,2H);13C NMR (125 MHz,DMSO)δ為:154.3(O—C),131.8(C—C),130.5(Ar—C),126.8(Ar—C),21.3(Me),15.8(Me);m/z=289.3。
按相同方法,改變苯酚衍生物,分別合成Cat.2~Cat.5,各催化劑表征結果如下。
淺黃色固體Cat.2(C7H7OTiCl3,分子量261.3),收率97.3%;XPS元素分析:C fund 33.0 cal.32.1,Ti fund 16.8 cal.18.2,Cl fund 38.7 cal.40.8;1H NMR(400 MHz,DMSO)δ為:2.19(CH3,s,3H),6.90(CH,d,2H),6.79(CH,d,2H);13C NMR(125 MHz,DMSO)δ為:155.7(O—C),132.1(C—C),129.4(Ar—C),115.9(Ar—C),20.6(Me);m/z=261.3。
黃色固體Cat.3(C6H4FOTiCl3,分子量265.3),收率95.3%;XPS元素分析:C fund 28.2 cal.27.1,F fund 6.9 cal.7.1,Ti fund 16.9 cal.18.0,Cl fund 38.8 cal.40.1;1H NMR(400 MHz,DMSO)δ為:6.97(CH,d,2H),6.79(CH,d,2H);13C NMR(125 MHz,DMSO)δ為:156.1(F—C),153.1(O—C),118.1(Ar—C),115.8(Ar—C);m/z=265.3。
復雜的菌類植物在潮濕的環境下野蠻生長。它們冒頭在林地表面,看起來不起眼,卻吸引了大量戶外攝影師的目光。
褐色固體Cat.4(C6H3F2OTiCl3,分子量283.3),收率97.0%;XPS元素分析:C fund 26.2 cal.25.4,F fund 12.8 cal.13.4,Ti fund 16.1 cal.16.8,Cl fund 36.8 cal.37.6;1H NMR(400 MHz,DMSO)δ:6.99(CH,d,2H),6.77(CH,d,H);13C NMR(125 MHz,DMSO)δ為:156.4(F—C),135.9(O—C),122.8(Ar—C),110.8(Ar—C);m/z=283.3。
褐黑色固體Cat.5(C6H2F3OTiCl3,分子量301.3),收率95.2%;XPS元素分析:C fund 24.6 cal.23.9,F fund 18.5 cal.18.9,Ti fund 15.3 cal.15.9,Cl fund 34.2 cal.35.3;1H NMR(400 MHz,DMSO)δ為:7.11(CH,s,H);13C NMR(125 MHz,DMSO)δ為:157.3(F—C),156.9(F—C),130.7(O—C),99.8(Ar—C);m/z=301.3。
1.4 減阻劑合成
采用低溫常壓液相本體法合成減阻劑。取1個250 mL Schlenk反應瓶,氮氣置換4~6次,使用熱風槍烘烤15 min,待冷卻后充氮氣。取1個玻璃連接器與干燥的安瓿瓶連接,氮氣置換4~5次,使用熱風槍烘烤15 min,待冷卻后在氮氣氛圍下稱取一定量的催化劑加入安瓿瓶中,在氮氣氛圍下將催化劑加入Schlenk反應瓶中。用注射器取MAO加入Schlenk反應瓶中,室溫下烷基化30 min。按n(1-辛烯)∶n(Ti)= 2 000:1的投料比加入1-辛烯單體。第1階段在0 ℃下聚合24 h,第2階段在5 ℃反應144 h。之后采用異丙醇將催化劑體系失活、過濾,得到的聚合物于真空烘箱中50 ℃下干燥至恒重,稱重計算單體轉化率及產物收率。
2 結果與討論
2.1 催化劑結構的影響
催化劑結構對1-辛烯聚合行為的影響見表1。Cat.1~Cat.5的主要區別是合成原料分別為2,4,6-三甲基苯酚、4-甲基苯酚、4-氟苯酚、2,6-二氟苯酚和2,4,6-三氟苯酚。從表1可看出,當催化劑配體上的推電子基團數量增多,單體轉化率與聚合物的Mη降低;當取代基為吸電子基團,或吸電子基團增多時,均能實現配體與過渡金屬Ti原子間化學鍵極性的加強,單體轉化率與聚合物Mη均相對提高,其中以2,6-二氟苯酚為原料的Cat.4和以2,4,6-三氟苯酚為原料的Cat.5具有理想的單體轉化率(大于96.9%),所得聚合物的Mη大于3.55×106。實驗結果表明,通過調整催化劑配體上取代基的吸電子能力可以調控催化劑活性位點的電子效應與配位環境,進而調控催化劑的催化性能,獲得超高分子量聚合物,即隨著催化劑配體上取代基吸電子能力增強或吸電子取代基數量增加,催化劑中Ti原子周圍的電子云密度降低,電正性增大,更易與由于極化而帶負電性的a-烯烴單體雙鍵的1-碳原子結合,發生配位、1,2-插入和鏈增長,從而獲得更高分子量的聚合物。由于烯烴單體帶負電性的碳原子更易與正電性大的Ti原子結合及配位,因此在相同聚合時間內單體轉化率更高、催化劑活性更高。考慮到2,4,6-三氟苯酚的價格比2,6-二氟苯酚的高,因此優選Cat.4 探討聚合條件對1-辛烯聚合行為的影響。
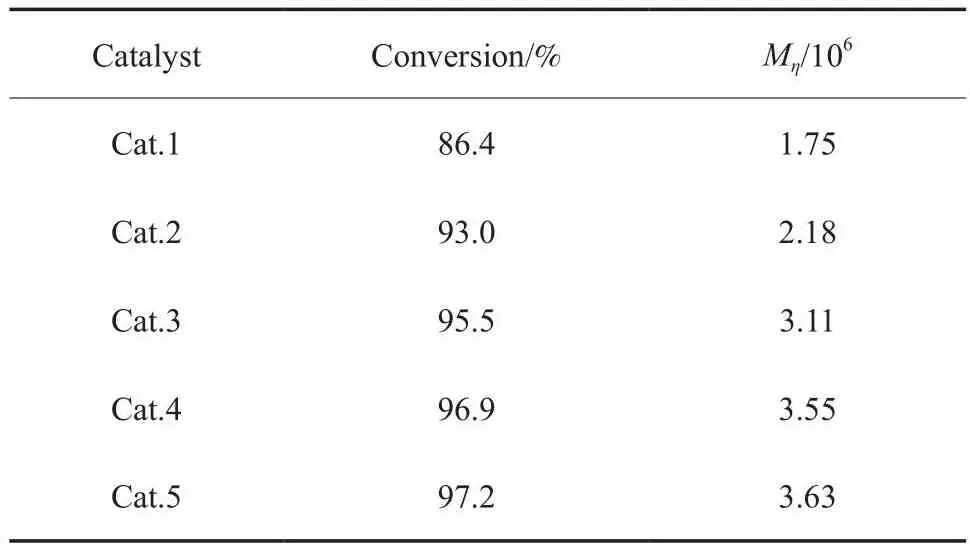
表1 催化劑結構對1-辛烯聚合行為的影響Table 1 Effects of catalyst structure on polymerization behaviour of 1-octene
2.2 聚合條件的影響
2.2.1 溫度的影響
第1階段聚合溫度對1-辛烯聚合的影響見表2。由表2可知,第1階段聚合溫度從-10 ℃升至0 ℃時,1-辛烯轉化率逐漸升高,聚合物的Mη增大。可能是由于溫度適當提高,會使聚合體系的黏度(1-辛烯液相本體聚合)降低,有利于單體布朗運動,從而提高聚合速率。即第1階段聚合溫度的略微升高有利于1-辛烯聚合,但當第1階段的聚合溫度從0 ℃升至25℃時,1-辛烯轉化率和聚合物Mη降低,說明第1階段的聚合溫度較高時,不能獲得超高分子量聚1-辛烯,可能是溫度較高引起了鏈轉移反應。
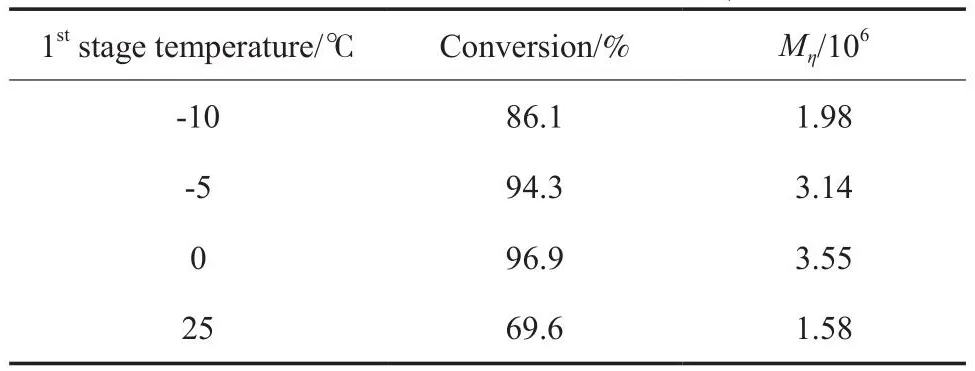
表2 第1階段聚合溫度對1-辛烯轉化率和聚合物Mη的影響Table 2 Effects of 1st stage polymerization temperature on 1-octene conversion and polymer Mη
2.2.2 聚合時間的影響
1-辛烯在第2階段的聚合時間對單體轉化率和聚合物Mη的影響見表3。由表3可知,隨第2階段聚合時間的延長,1-辛烯的轉化率增大。但當聚合時間達到144 h后,1-辛烯的轉化率趨于平穩。聚合時間144 h 后聚合體系成為了塊狀固體,未反應的單體傳質較難,故難以發生聚合反應。因此,聚合時間144 h 后不宜再延長反應時間。聚合物Mη隨第2階段聚合時間的延長而增大,即反應時間從144 h延至168 h,聚合物的Mη仍在增大。說明Cat.4/MAO催化劑體系的穩定性較好。
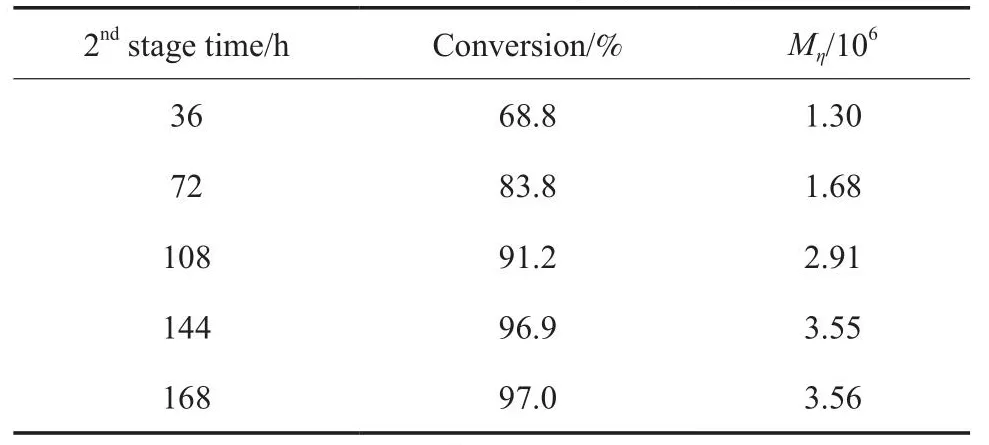
表3 第2階段聚合時間對1-辛烯轉化率和聚合物Mη的影響Table 3 Effects of 2nd stage polymerization time on 1-octene conversion and polymer Mη
2.2.3 助催化劑種類及鋁鈦摩爾比的影響
助催化劑對1-辛烯轉化率及聚合物Mη的影響見表4。由表4可看出,MAO是最適宜的助催化劑。與AlEt3和AlEt2Cl比較,采用Cat.4/MAO催化體系催化1-辛烯液相本體聚合,1-辛烯的轉化率最高,聚合物Mη最大;采用AlEt2Cl作助催化劑的聚合效果最差。
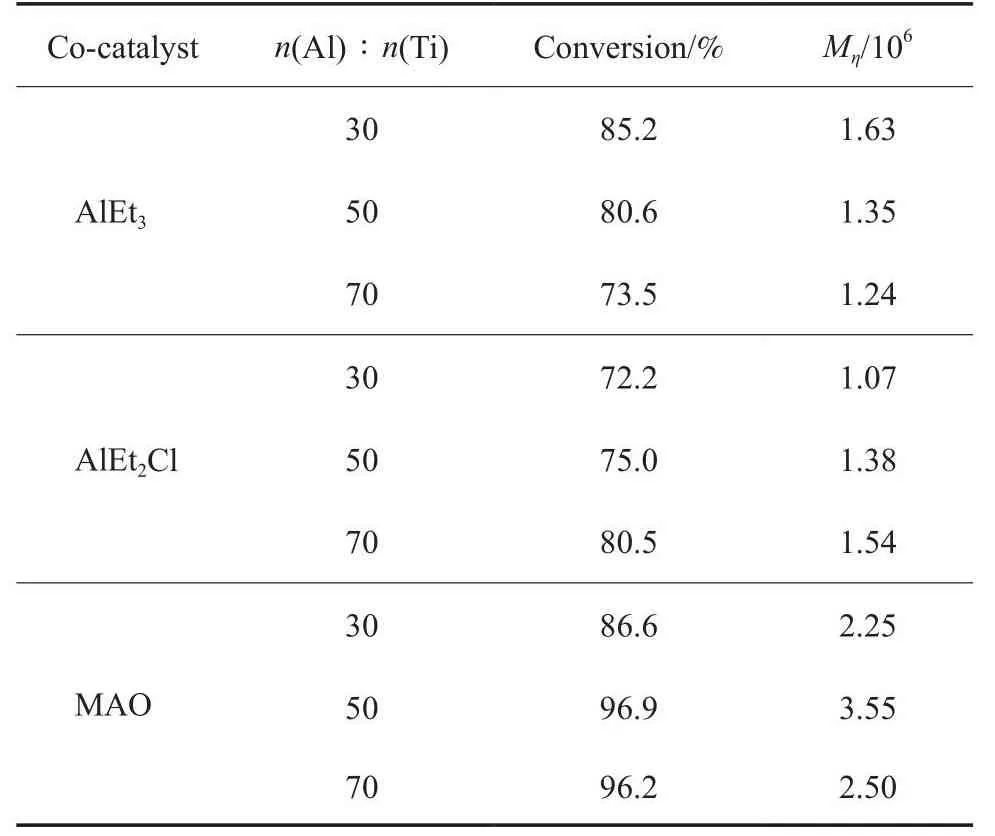
表4 助催化劑對1-辛烯轉化率和聚合物Mη的影響Table 4 Effects of co-catalyst on 1-octene conversion and polymer Mη
助催化劑不同,穩定存在的形式也不同。MAO雖是三甲基鋁(TMA)的水解產物,具有Al(Me2)O重復結構的低聚物,但真正起烷基化作用、鏈轉移作用的是MAO中存在的少量TMA。由于TMA反應迅速,與催化劑活性中心作用進行烷基化反應也迅速。MAO的另一作用是結合并穩定從主催化劑上解離下來的氯離子形成絡合反離子,有利于活性中心的穩定。MAO的Al(Me2)O結構也可能有利于形成1-辛烯單體與催化劑活性位點配位、插入、鏈增長的環境。
由表4還可看出,AlEt3為助催化劑時,1-辛烯的轉化率和聚合物Mη隨AlEt3用量的增加而降低。這可能是由于,隨AlEt3用量的增加,催化劑中的氧化鈦(Ⅳ)被過度還原成不適宜1-辛烯聚合的、更低價的Ti,如Ti(Ⅱ)等[25-26],導致催化劑活性降低,因此,1-辛烯的轉化率降低。AlEt3的加入量增加,聚合物增長鏈向AlEt3發生鏈轉移反應的概率增加,導致聚合物Mη降低。
與AlEt3比較,由于AlEt2Cl中含有電負性較強的Cl,使Al原子顯較強的正電性(即Al原子周圍的電荷會偏向Cl),Al與Et的結合強度增加,因此,AlEt2Cl中的Et較難失去。AlEt2Cl 與Cat.4組成催化劑體系,催化1-辛烯聚合的結果與AlEt3的不同。由表4可看出,AlEt2Cl 為助催化劑時,1-辛烯的轉化率和聚合物Mη隨AlEt2Cl加入量的增加而增加。但聚合效果比MAO作助催化劑差。
綜上所述,1-辛烯聚合適宜的條件為:催化體系Cat.4/MAO;第1階段聚合溫度為0 ℃,聚合時間24 h;第2階段聚合溫度為5 ℃,聚合時間144 h;n(Al)∶n(Ti)=50∶1,n(1-辛烯)∶n(Ti)=2 000∶1。在該條件下,單體轉化率為96.9%,產物聚1-辛烯的Mη為3.55×106。
2.3 聚合物結構與性能表征
對優化條件下制備的聚1-辛烯進行結構與性能表征。
2.3.1 FTIR表征結果
聚1-辛烯的FTIR譜圖見圖1。
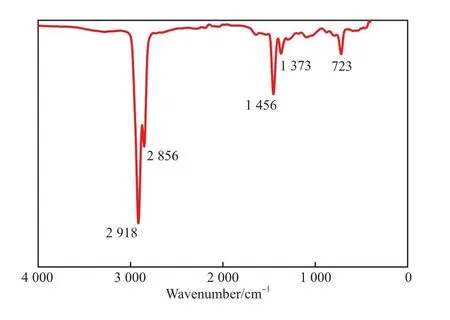
圖1 聚1-辛烯的FTIR譜圖Fig.1 FTIR spectrum of poly(1-octene).
從圖1可看出,2 856 cm-1處的吸收峰對應CH2的C—H鍵伸縮振動;2 918 cm-1處的吸收峰對應CH3的C—H鍵伸縮振動;1 456 cm-1處的吸收峰對應CH2的剪切和不對稱振動;1 373 cm-1處的吸收峰為C—H鍵的彎曲振動;723 cm-1處的吸收峰為CH2搖擺振動;772 cm-1處的吸收峰歸屬于附著于主鏈上的—(CH2)—CH3側基。上述吸收峰均對應于聚合物主鏈和側基的飽和C—C單鍵。在圖1中未出現分別對應=C—H鍵振動、伸縮振動及C=C鍵伸縮振動的990,3 080,1 640 cm-1處的吸收峰。可能是由于聚合物的分子量超高,因β-H鏈轉移產生的C=C鍵鏈末端占比極低,FTIR無法識別。
2.3.21H NMR表征結果
聚1-辛烯的1H NMR譜圖見圖2。如圖2所示,在δ=1.68,1.40,0.95處的吸收峰分別歸屬于聚1-辛烯CH,CH2,CH3中的H原子。圖中未出現CH=CH中氫原子的信號峰,說明所得聚1-辛烯分子鏈中雙鍵含量極低。
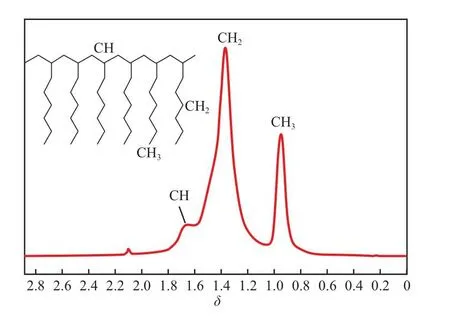
圖2 聚1-辛烯的1H NMR譜圖Fig.2 1H NMR spectrum of poly(1-octene).
聚1-辛烯的13C NMR譜圖見圖3。由圖3可知,δ=41.2,35.8,33.3,32.5,30.5,27.4,23.2,14.2分別對應聚1-辛烯分子鏈上1#~8#碳原子的信號峰,是1-辛烯發生“頭-尾”相連的聚合物分子鏈信號峰。在δ=39.5處沒有信號峰,表明不存在由于主鏈“頭-頭”相連形成的兩個直接相連的次甲基。6#碳原子的信號峰也未分裂成δ=27.4,27.6兩個信號峰,說明不存在由于“尾-尾”相連形成的兩個直接相連的亞甲基。未出現對應于C=C鍵的信號峰,說明所得聚1-辛烯分子鏈中雙鍵含量可忽略。

圖3 聚1-辛烯的13C NMR譜圖Fig.3 13C NMR spectrum of poly(1-octene).
2.3.3 WAXD分析
聚1-辛烯熱壓膜的WAXD 譜圖見圖4。從圖4可看出,在2θ=7°,20°附近出現的兩個寬峰,是聚1-辛烯的特征衍射峰。未出現尖銳的衍射峰,表明由Cat.4合成的聚1-辛烯是無定形結構,不結晶。具有無定形結構的聚1-辛烯作為油品管道輸送用減阻劑較容易溶于油品中,易于處理。
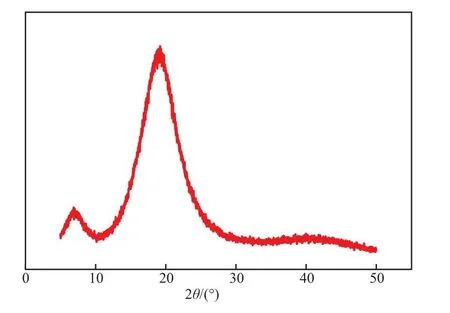
圖4 聚1-辛烯的WAXD譜圖Fig.4 XRD pattern of poly(1-octene).
2.4 減阻效果分析
按 SY/T 6578—2016[24]規定的方法,采用室內測試環道開展了減阻性能測試,在測試減阻聚合物溶液前,先測定0#柴油在相同溫度下流經管道因摩擦而產生的壓降。加有5 mg/kg減阻劑的0#柴油在相同的管道上進行減阻測試,得到壓降,按式(2)計算減阻率(DR)。
式中,Δp0為基礎測試條件下管段摩阻壓降,kPa;ΔpDRA為減阻率測試條件下管段摩阻壓降,kPa。
計算結果表明,本工作制備的聚1-辛烯的減阻率為46.9%。在相同條件下測試了幾種國產減阻劑的減阻率,減阻率最高的為43.5%;美國Conoco公司的減阻劑Liquid Power的減阻率為47.2%。說明由Cat.4/MAO體系制備的減阻劑,減阻效果與Liquid Power產品相當。
3 結論
1)在活性組分TiCl4中引入含F取代基的芳氧基基團,可調控Ti原子周圍的電子效應與配位環境,提高催化劑活性中心的穩定性。優選含2,6-二氟苯氧基的Cat.4催化1-辛烯聚合獲得超高分子量聚合物。
2)Cat.4/MAO體系催化1-辛烯聚合適宜的條件為:第1階段聚合溫度0 ℃,聚合時間24 h;第2階段聚合溫度5 ℃,聚合時間144 h;n(Al)∶n(Ti)=50∶1,n(1-辛烯)∶n(Ti)=2 000∶1。在該條件下,單體轉化率為96.9%,聚1-辛烯的Mη高達 3.55×106。
3)所得聚1-辛烯為無定形結構,易溶于油品中,減阻率為46.9%,優于國內其他油溶性減阻劑,與進口產品性能相當。
4)催化劑制備和1-辛烯聚合均在溫和溫度和常壓下進行,成本低。而且在聚1-辛烯作為減阻劑處理及使用過程中都沒有CO2的產生和排放,符合國家雙碳戰略目標。
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
