超高效液相色譜-串聯(lián)質(zhì)譜法測定調(diào)味料中嗎啡、可待因、蒂巴因、那可丁和罌粟堿殘留量
2023-03-20 08:56:14張哲張瑩趙桐趙靈智沈晶萍王世琨李思圓
中國調(diào)味品 2023年3期
張哲,張瑩,趙桐,趙靈智,沈晶萍,王世琨,李思圓
(吉林農(nóng)業(yè)大學 生命科學學院,長春 130118)
罌粟殼中含有嗎啡、可待因、罌粟堿、那可丁、蒂巴因等生物堿,因其具有鎮(zhèn)靜、止痛、成癮、損害神經(jīng)系統(tǒng)等特性被列為毒品,嚴禁將其添加到食品中。由于火鍋底料中添加罌粟殼會使火鍋味道更美、口感更足,食用后容易上癮,有些不良商家將罌粟殼加入火鍋調(diào)料中以吸引回頭客來牟取利益[1]。國家早在2008年印發(fā)了《食品中可能違法添加的非食用物質(zhì)和易濫用的食品添加劑品種名單(第一批)》,規(guī)定火鍋中不得違法添加罌粟殼,2011年又將禁用的食品類別擴大到火鍋底料及小吃。歷年發(fā)布的《國家食品安全抽檢實施細則》中更是明確將嗎啡、可待因、罌粟堿、那可丁、蒂巴因列為餐飲食品自制火鍋調(diào)味料(底料、蘸料)的日常監(jiān)測項目。
目前食品中罌粟殼成分的檢測方法主要有氣相色譜法[2]、液相色譜法、氣相色譜/質(zhì)譜法、液相色譜-質(zhì)譜/質(zhì)譜法。其中液相色譜-質(zhì)譜/質(zhì)譜法因具有分辨率高、靈敏度高、檢測限低,定性、定量準確,可進行痕量分析等特點,已被市場監(jiān)管總局指定為檢測食品中嗎啡、可待因、罌粟堿、那可丁和蒂巴因的補充檢驗方法,成為檢測罌粟殼類生物堿的主要方法[3-4]。在色譜柱選擇上,由于嗎啡等生物堿極性大,在普通C18反相柱上保留時間短、峰形差,達不到較好的分離效果[5],大多采用BEH HILIC色譜柱進行檢測[6-10]。然而HILIC柱為正向色譜柱,使用溶劑有限,只能使用70%以上乙腈,難以預平衡,且價格昂貴、使用壽命短,實驗室普及率不高[11-12]。在樣品前處理上,QuEChERS凈化法操作簡單,但凈化效果不理想,且對嗎啡等有強吸附,回收率低[13-16],常采用陽離子交換固相萃取凈化法[17-18],而陽離子交換固相萃取柱除油脂效果不理想,操作較繁瑣,且容易發(fā)生小柱堵塞現(xiàn)象[19-20],無法實現(xiàn)批量檢測。
本試驗采用實驗室常用的Kinetex?2.6 μm Biphenyl 100 ?反相色譜柱分離,建立超高效液相色譜-串聯(lián)質(zhì)譜法測定調(diào)味料中罌粟殼類生物堿,嗎啡等5種生物堿得到了很好的色譜保留和分離,且峰形對稱;在樣品處理上,樣品經(jīng)乙腈提取、鹽析、低溫高速離心后,將提取液氮吹近干,用10%乙腈溶液(含0.1%甲酸)溶解殘渣后,再經(jīng)低溫高速離心、過膜除去樣品中干擾物,獲得了理想的凈化、回收效果,使樣品前處理過程更加簡便,適合大批量的日常分析檢測。
1 材料與方法
1.1 材料
調(diào)味料:固態(tài)火鍋底料、半固態(tài)米線調(diào)味醬、半固態(tài)小龍蝦調(diào)味醬、湯料共4種,購于超市。
自制火鍋調(diào)味料(蘸料):海鮮汁、沙茶醬、孜然油、麻油、辣椒油、芝麻油共6種,購于火鍋店。
1.2 試劑
甲醇中嗎啡標準溶液(M-005-1ML,濃度1 mg/mL)、甲醇中可待因標準溶液(C-006-1ML,濃度1 mg/mL)、甲醇中蒂巴因標準溶液(T-115-1ML,濃度1 mg/mL)、甲醇中嗎啡-D3標準溶液(M-006-1ML,濃度1 mg/mL)、甲醇中可待因-D3標準溶液(C-007-1ML,濃度1 mg/mL):Cerilliant;鹽酸罌粟堿標準物質(zhì)(純度99.9%,CCHM700898)、那可丁標準物質(zhì)(純度97%,CCHM700909):CATO Research Chemicals Inc.;甲醇(色譜純)、乙腈(色譜純):Fisher Scientific公司;甲酸(色譜純):上海安譜實驗科技股份有限公司;無水乙酸鈉(分析純):天津市致遠化學試劑有限公司;無水硫酸鎂(分析純):北京邁瑞達科技有限公司;0.1%甲酸-水溶液(體積比);0.1%甲酸-甲醇溶液(體積比);10%乙腈-水溶液(含0.1%甲酸);針頭過濾器(0.2 μm,13 mm):Pall Corporation。
1.3 主要儀器與設備
API4000+超高效液相色譜-串聯(lián)質(zhì)譜聯(lián)用儀(ESI離子源) 美國AB SCIEX公司;Kinetex?2.6 μm Biphenyl 100 ?色譜柱(100 mm×3.0 mm) 美國Phenomenex公司;渦旋混合器(重載數(shù)顯型) 美國Talboys公司;KQ-500DE超聲波清洗器 江蘇昆山市超聲儀器有限公司;KH20R-Ⅱ高速冷凍離心機 湖南凱達科學儀器有限公司;TTL-DC氮吹儀 北京同泰聯(lián)科技發(fā)展有限公司。
1.4 液相色譜-質(zhì)譜/質(zhì)譜條件
1.4.1 色譜條件
色譜柱:Kinetex?2.6 μm Biphenyl 100 ?(100 mm×3.0 mm);柱溫:40 ℃;流速:0.4 mL/min;進樣量:5 μL;流動相:A相為 0.1%甲酸-水(體積比),B相為0.1%甲酸-甲醇(體積比)。流動相及梯度洗脫條件見表1。
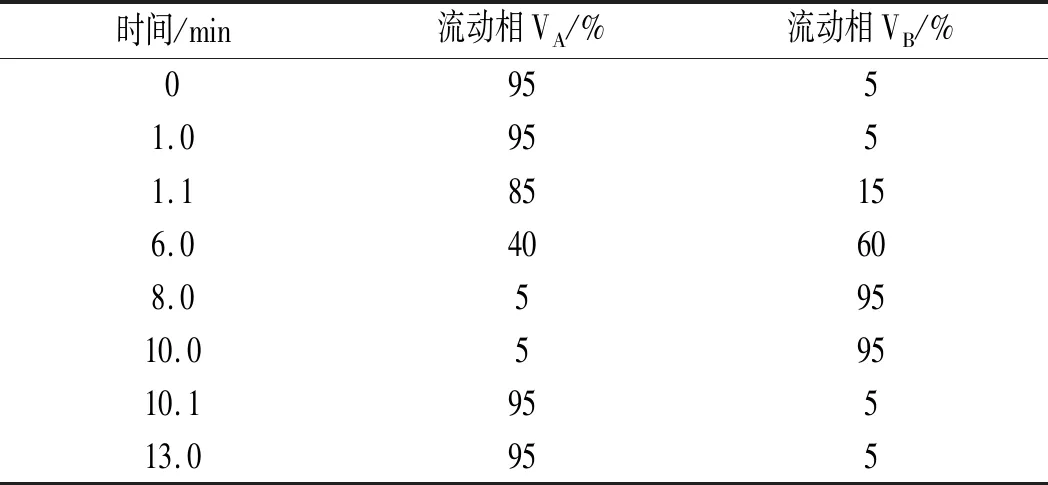
表1 流動相及梯度洗脫條件Table 1 Mobile phases and gradient elution conditions
1.4.2 質(zhì)譜條件
離子源:ESI;掃描方式:正離子模式,多反應監(jiān)測(MRM);離子源參數(shù):電噴霧電壓(IS):5 500 V;霧化氣壓力(GS1):55 psi;輔助氣壓力(GS2):60 psi;氣簾氣壓力(CUR):30 psi;離子源溫度(TEM):550 ℃;噴撞氣(CAD):10 mL/min。監(jiān)測離子對、碰撞氣能量和去簇電壓參數(shù)見表2。

表2 監(jiān)測離子對、碰撞氣能量和去簇電壓參數(shù) Table 2 Monitoring ion pair,collision gas energy and declustering potential parameters
1.5 標準溶液配制
1.5.1 標準儲備液配制
精密稱取罌粟堿、那可丁標準品各10 mg(精確至0.01 mg),分別置于10 mL容量瓶中,加0.5%甲酸-甲醇溶液溶解并定容至刻度,罌粟堿、那可丁濃度各1.0 mg/mL。嗎啡、可待因、蒂巴因、嗎啡-D3、可待因-D3均為購置現(xiàn)成的標準溶液,其濃度均為1.0 mg/mL。
1.5.2 混合標準溶液配制
準確吸取嗎啡、可待因標準儲備液各100 μL,蒂巴因標準儲備液20 μL,那可丁、罌粟堿標準儲備液各10 μL于10 mL容量瓶中,用乙腈稀釋并定容于10 mL。配制的混合標準溶液中嗎啡、可待因濃度為10 μg/mL,蒂巴因濃度為2 μg/mL,那可丁、罌粟堿濃度為1 μg/mL。
1.5.3 混合標準使用液配制
準確吸取混合標準溶液0.1 mL于10 mL容量瓶中,用20%乙腈水溶液定容至刻度。配制的混合標準使用液中嗎啡、可待因濃度為100 ng/mL,蒂巴因濃度為20 ng/mL,那可丁、罌粟堿濃度為10 ng/mL。
1.5.4 混合內(nèi)標溶液配制
準確吸取嗎啡-D3、可待因-D3標準儲備液各10 μL于10 mL容量瓶中,用乙腈稀釋并定容于10 mL。配制的混合內(nèi)標溶液的濃度均為1 μg/mL。
1.5.5 混合內(nèi)標工作液配制
準確吸取混合內(nèi)標溶液1.0 mL于10 mL容量瓶中,用20%乙腈水溶液定容至刻度。配制的混合內(nèi)標工作液的濃度均為100 ng/mL。
1.5.6 混合標準工作溶液系列配制
分別準確吸取混合標準使用液5,10,20,50,100,200 μL于6個樣品瓶中,各加入100 ng/mL混合內(nèi)標工作液50 μL,用10%乙腈-水溶液(含0.1%甲酸)定容至1.0 mL,混勻。配制的混合標準工作溶液系列中嗎啡、可待因濃度分別為0.5,1.0,2.0,5.0,10.0,20.0 ng/mL;蒂巴因濃度分別為0.1,0.2,0.4,1.0,2.0,4.0 ng/mL;那可丁、罌粟堿濃度分別為0.05,0.1,0.2,0.5,1.0,2.0 ng/mL。此混合標準工作溶液中內(nèi)標濃度均為5.0 ng/mL。
1.6 樣品處理
稱取1.50 g(精確至0.01 g)試樣于50 mL離心管中,加入1 μg/mL混合內(nèi)標溶液75 μL,固體樣品加3 mL水,半固體樣品加2 mL水,液體樣品不加水,渦旋振蕩30 s,準確加入乙腈15 mL,渦旋1 min,超聲提取30 min,加入6 g無水硫酸鎂和1.5 g無水醋酸鈉粉末,立即渦旋振蕩2 min,吸附樣品中的全部水分,以9 500 r/min離心5 min(4 ℃),準確吸取1.0 mL上清液于10 mL濃縮管中,于40 ℃水浴中氮吹至近干,準確加入1.0 mL 10%乙腈溶液(含0.1%甲酸)溶解殘渣,渦旋1 min,超聲2 min,再渦旋1 min后,將溶液轉(zhuǎn)移至1.5 mL離心管中,以9 500 r/min離心5 min(4 ℃),上清液過0.22 μm濾膜后,進行超高效液相色譜-串聯(lián)質(zhì)譜測定。
2 結果與分析
2.1 色譜柱的選擇
以0.1%甲酸-水-0.1%甲酸-乙腈為流動相,考察了Kinetex?2.6 μm Biphenyl 100 ?(100 mm×3.0 mm)和Kinetex?2.6 μm F5 100 ?(50 mm×3.0 mm)兩款反相色譜柱對嗎啡、可待因、蒂巴因、那可丁和罌粟堿的分離效果,結果:在Kinetex?2.6 μm F5 100 ?色譜柱上,嗎啡岀峰快,保留時間短,峰形較寬、不對稱,響應值低,且5種生物堿各離子對的離子比不穩(wěn)定(見圖1),可見此款反相色譜柱不是首選柱,這與文獻[5]、文獻[20]的研究結果一致;而在Kinetex?2.6 μm Biphenyl 100 ?色譜柱上,嗎啡等5種生物堿均有合適的保留時間和令人滿意的峰形,且響應值增高(見圖2)。分析原因:Kinetex?2.6 μm Biphenyl 100 ?柱是一款核-殼聯(lián)苯基色譜柱,這種由核-殼顆粒帶來的高性能配合獨特的固定相,可以實現(xiàn)反相保留以及增強極性與芳香類化合物的選擇性,且能顯著改善色譜峰形,提高靈敏度,其色譜分離相互作用機理:由聯(lián)苯基雙環(huán)結構所產(chǎn)生的高密度電子云可起到類似弱陽離子交換的作用,使得堿性分析物的保留增強。
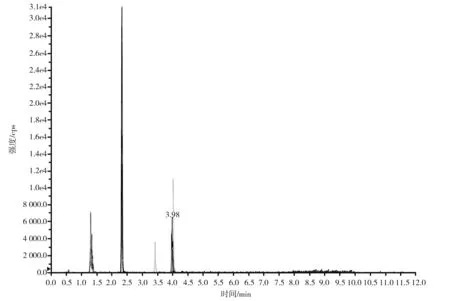
圖1 Kinetex? 2.6 μm F5 100 ?柱-混合標準工作溶液總離子流色譜圖Fig.1 The total ion current chromatogram of Kinetex? 2.6 μm F5 100 ? column-mixed standard working solution
圖2 Kinetex? 2.6 μm Biphenyl 100 ?柱-混合標準工作溶液總離子流色譜圖Fig.2 The total ion current chromatogram of Kinetex? 2.6 μm Biphenyl 100 ? column-mixed standard working solution
2.2 流動相的選擇
在Kinetex?2.6 μm Biphenyl 100 ?色譜柱(100 mm×3.0 mm)上分離,分別以0.1%甲酸-水和0.1%甲酸-乙腈,0.1%甲酸-水(含10 mmol/L甲酸銨)和0.1%甲酸-乙腈,0.1%甲酸-水和0.1%甲酸-甲醇為流動相,按表1中梯度程序洗脫,考察3種流動相對嗎啡、可待因、蒂巴因、那可丁和罌粟堿的分離效果,結果表明,以0.1%甲酸-水和0.1%甲酸-乙腈為流動相,那可丁和罌粟堿兩色譜峰重合(見圖2),在0.1%甲酸-水中加入10 mmol/L甲酸銨后,那可丁和罌粟堿兩峰不但沒有分開,5種生物堿的響應值反而下降。改用0.1%甲酸-水和0.1%甲酸-甲醇為流動相,5種生物堿均得到了良好的色譜分離,各離子對的離子比更穩(wěn)定,且嗎啡、可待因的響應值顯著提高(見圖3),故選用0.1%甲酸-水和0.1%甲酸-甲醇為流動相。
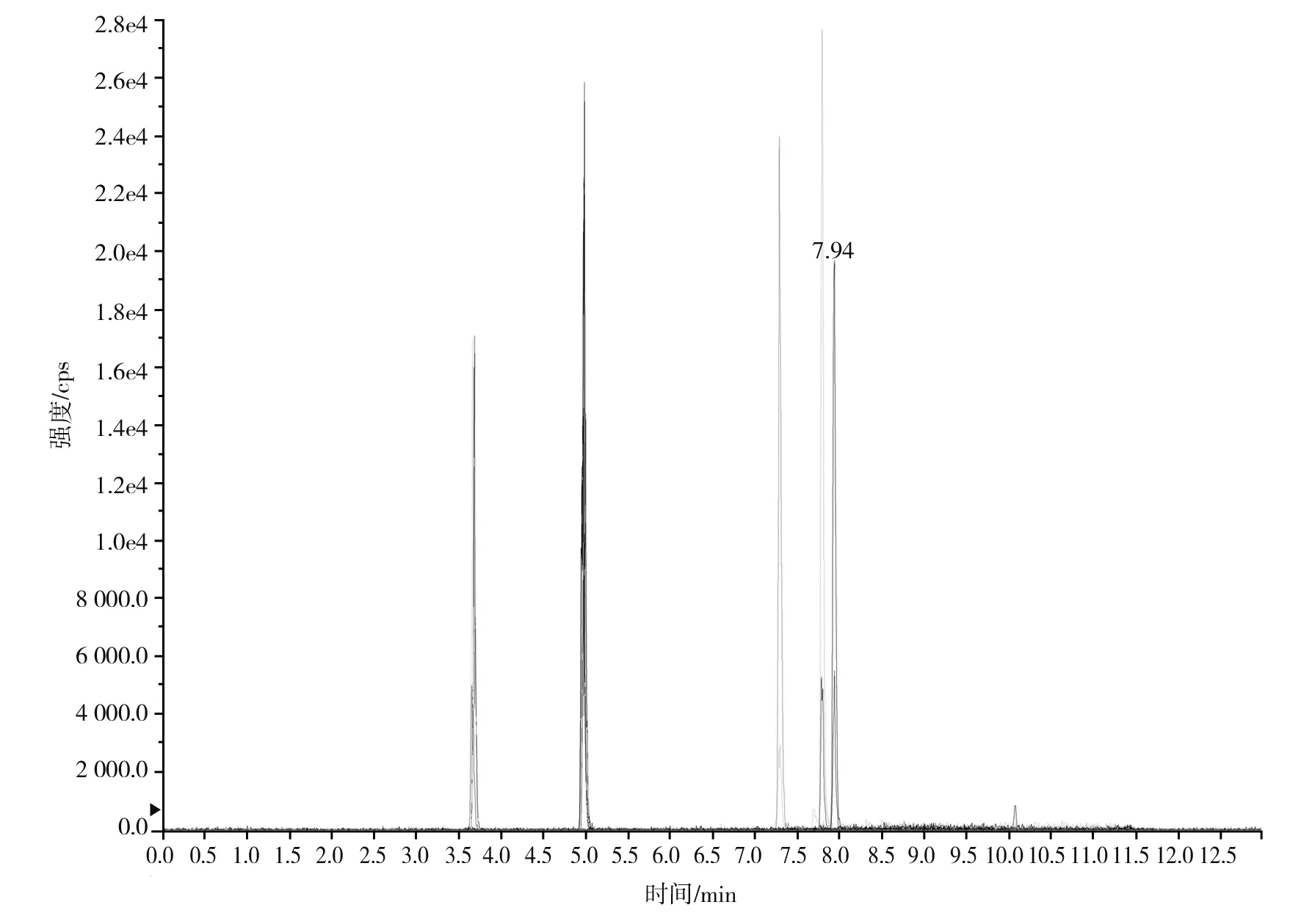
圖3 5種生物堿混合標準工作溶液的總離子流色譜圖Fig.3 The total ion current chromatogram of mixed standard working solution of five kinds of alkaloids
2.3 質(zhì)譜條件優(yōu)化
根據(jù)5種生物堿的化學性質(zhì),采用電噴霧電離正離子模式掃描。針泵進樣100 ng/mL標準溶液,5種生物堿離子化時均得到[M+H]+分子離子作為母離子。調(diào)節(jié)碰撞氣能量(CE)值,將母離子進一步碰碎,分別選擇響應值較高的兩個子離子,按多反應監(jiān)測(MRM)組建監(jiān)測離子對,用Ramp進一步優(yōu)化碰撞氣能量CE和去簇電壓DP參數(shù)(見表2)。設定離子源參數(shù)和液相色譜參數(shù),進樣分析混合標準工作溶液,驗證所選擇的離子對的響應值和穩(wěn)定性滿足檢測要求。5種生物堿混合標準工作溶液的多反應監(jiān)測(MRM)色譜圖見圖4。
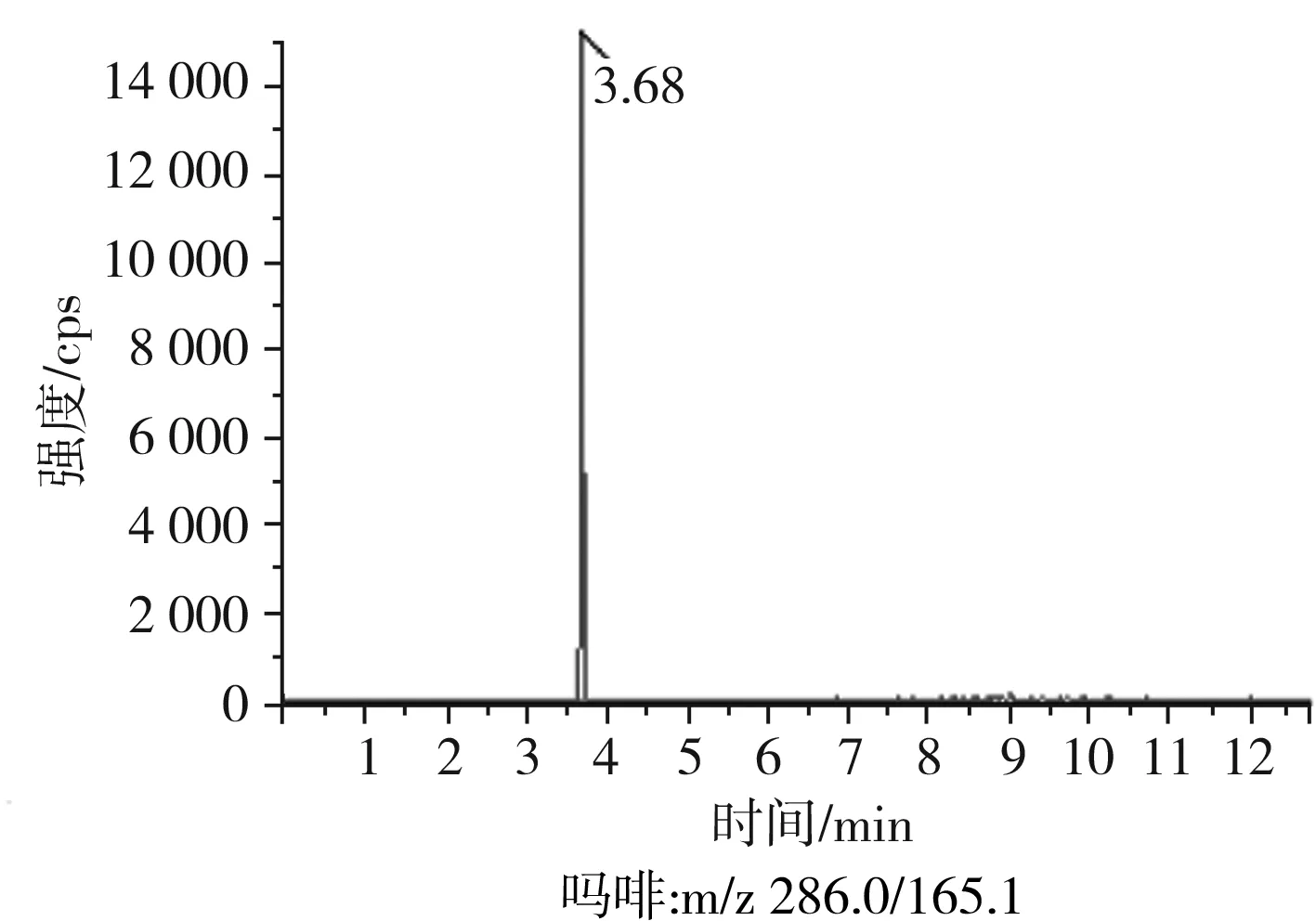
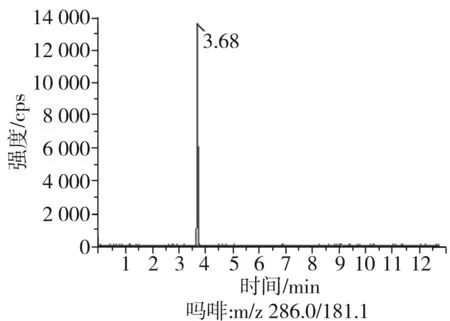
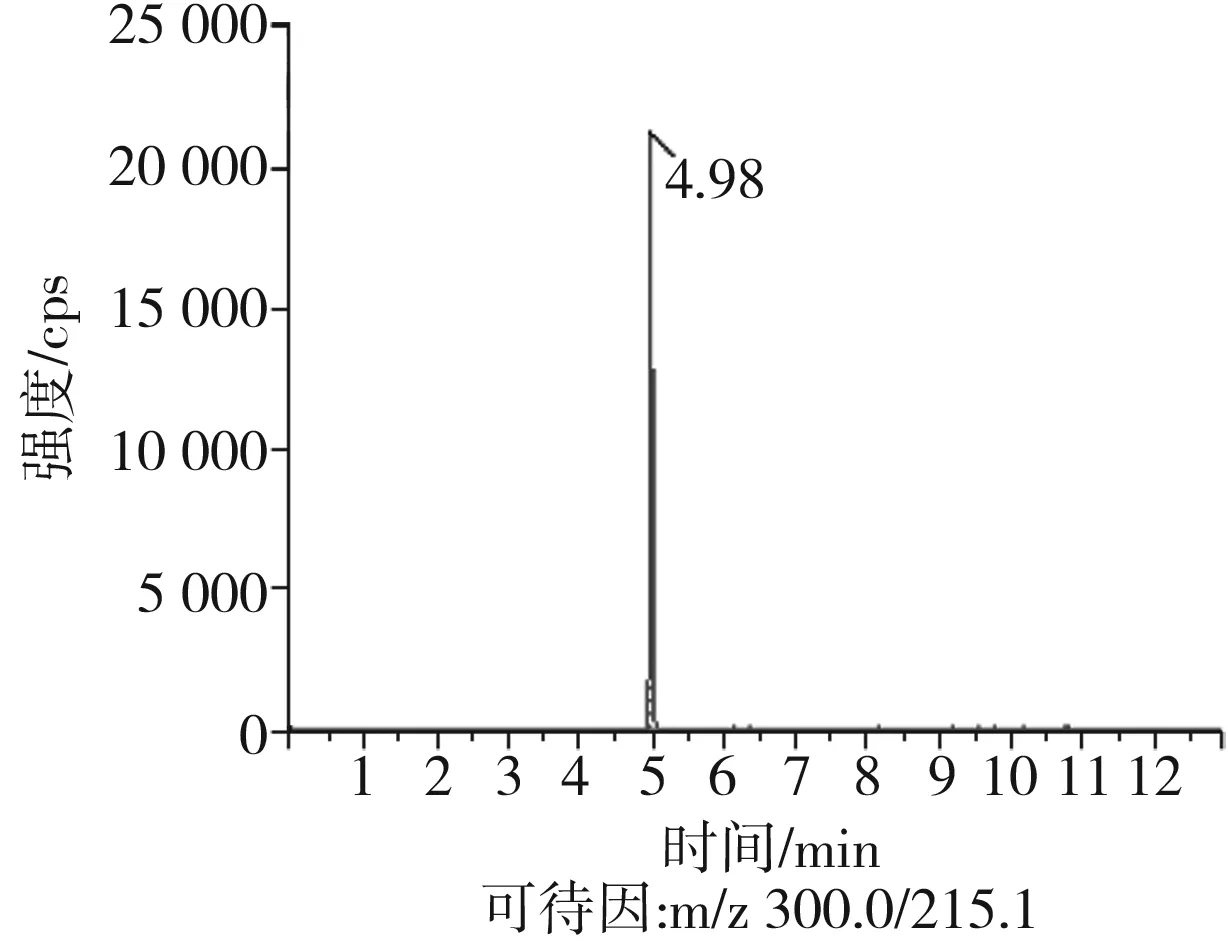
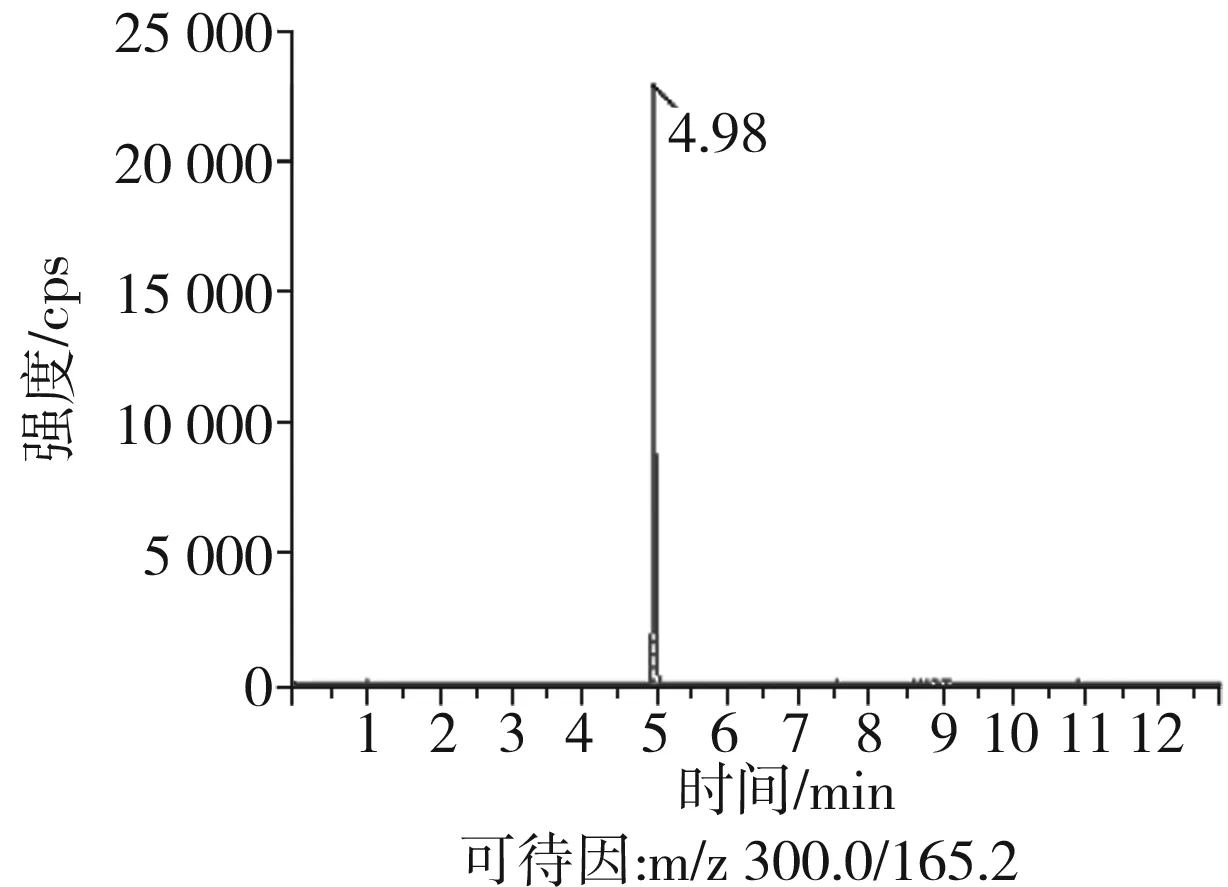
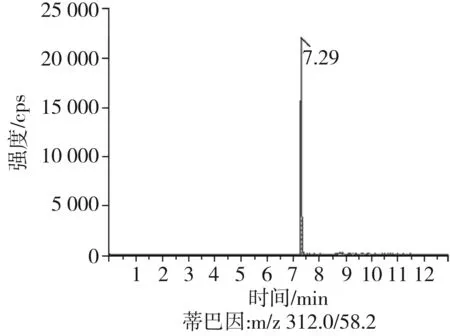
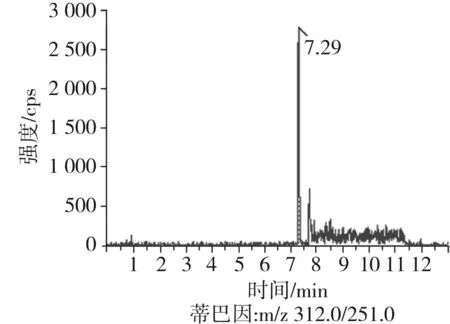
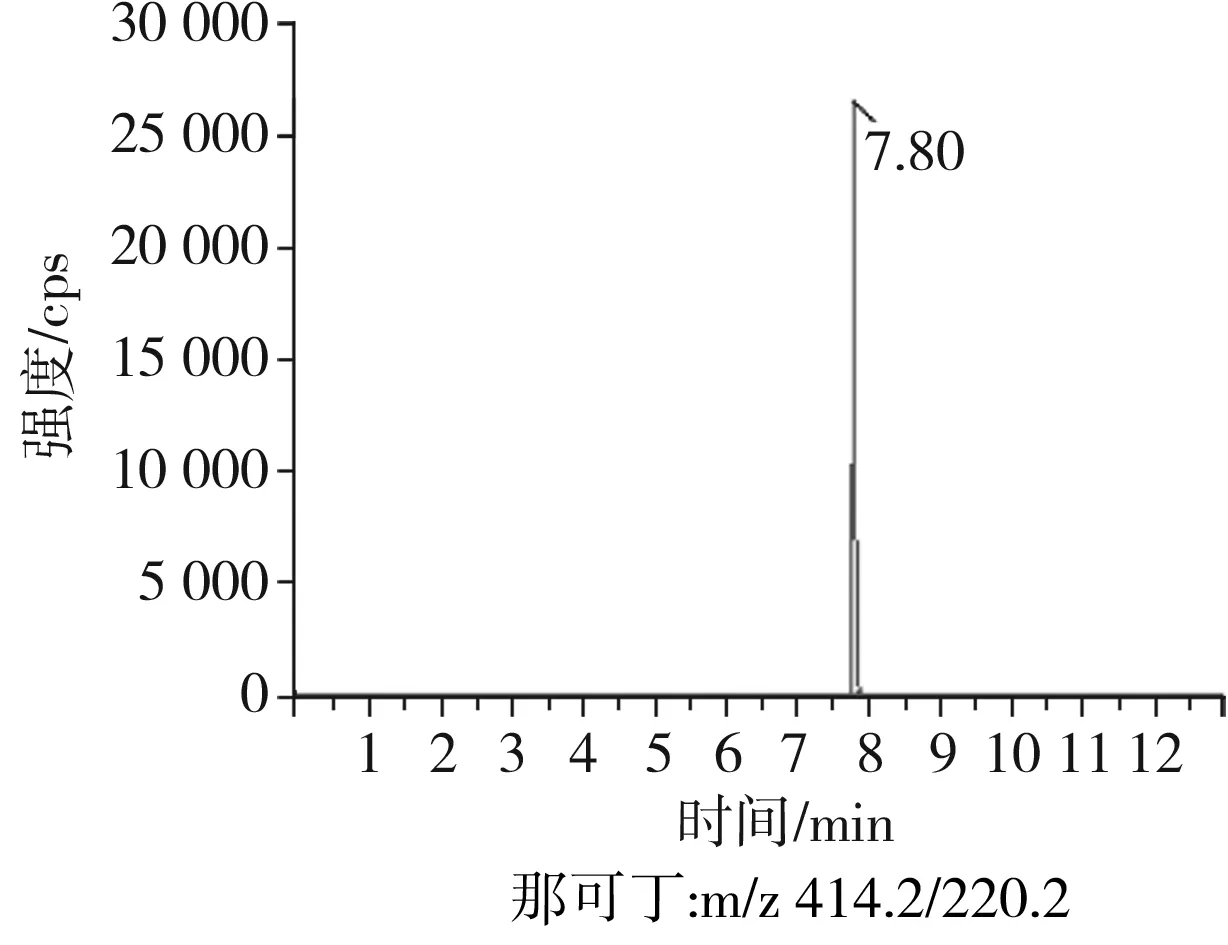
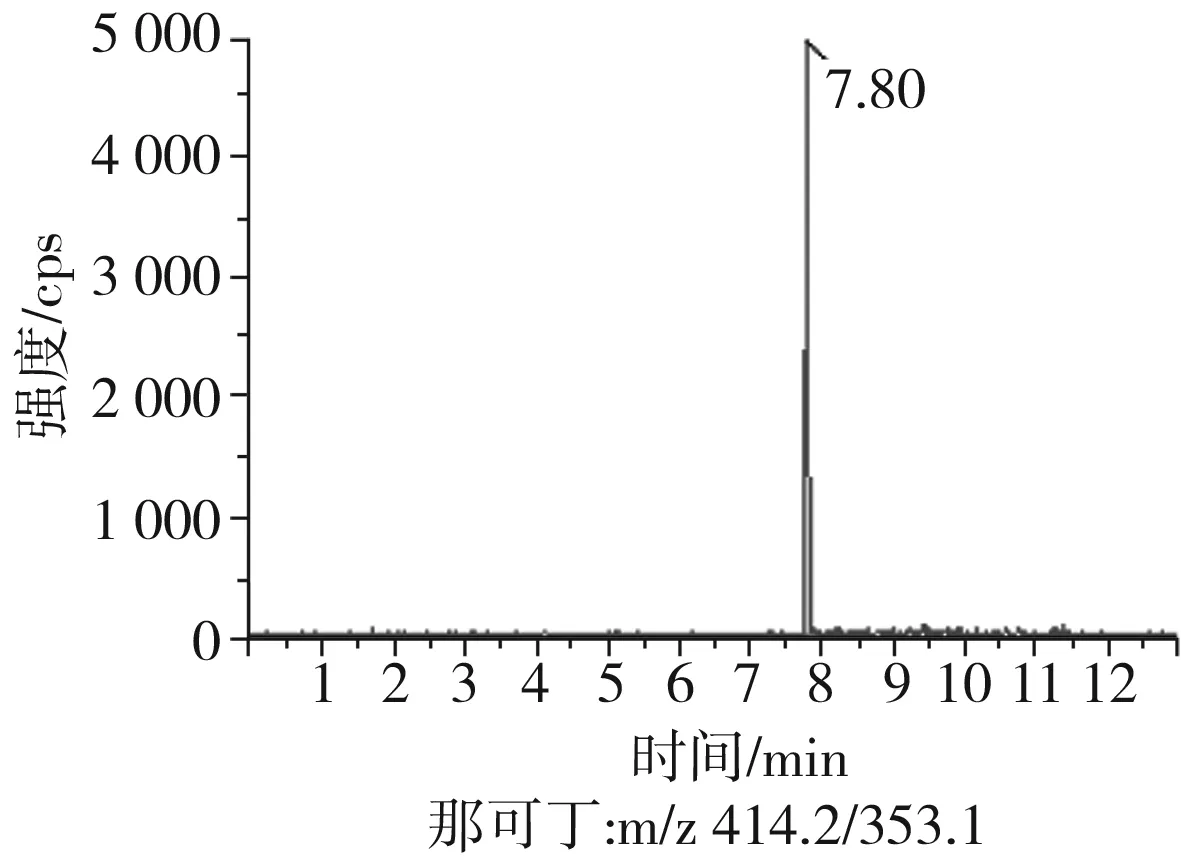
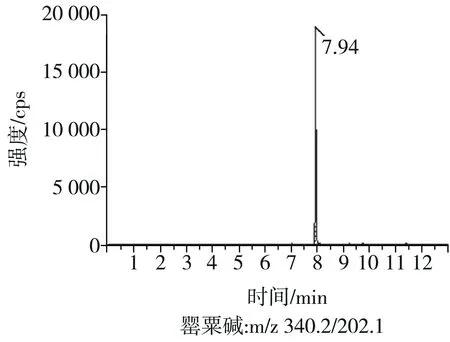
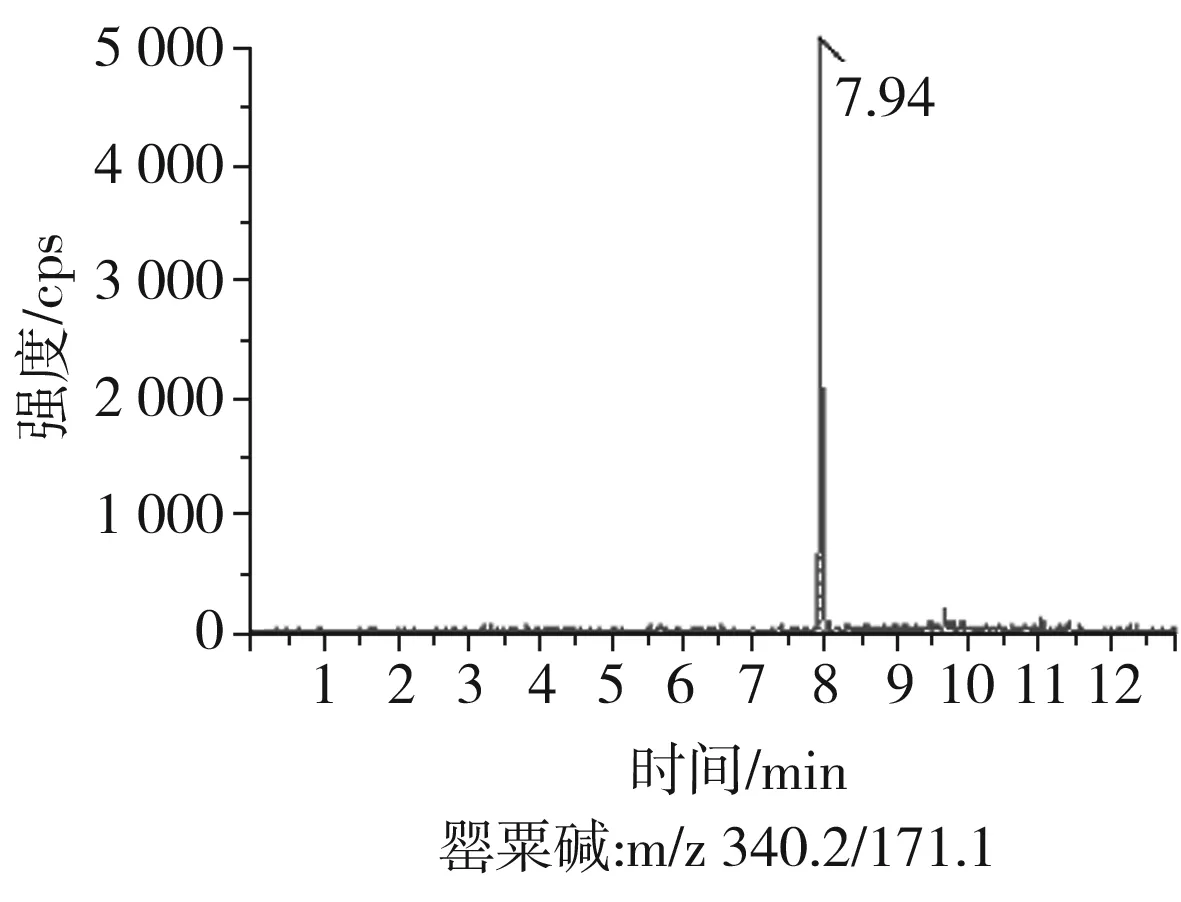
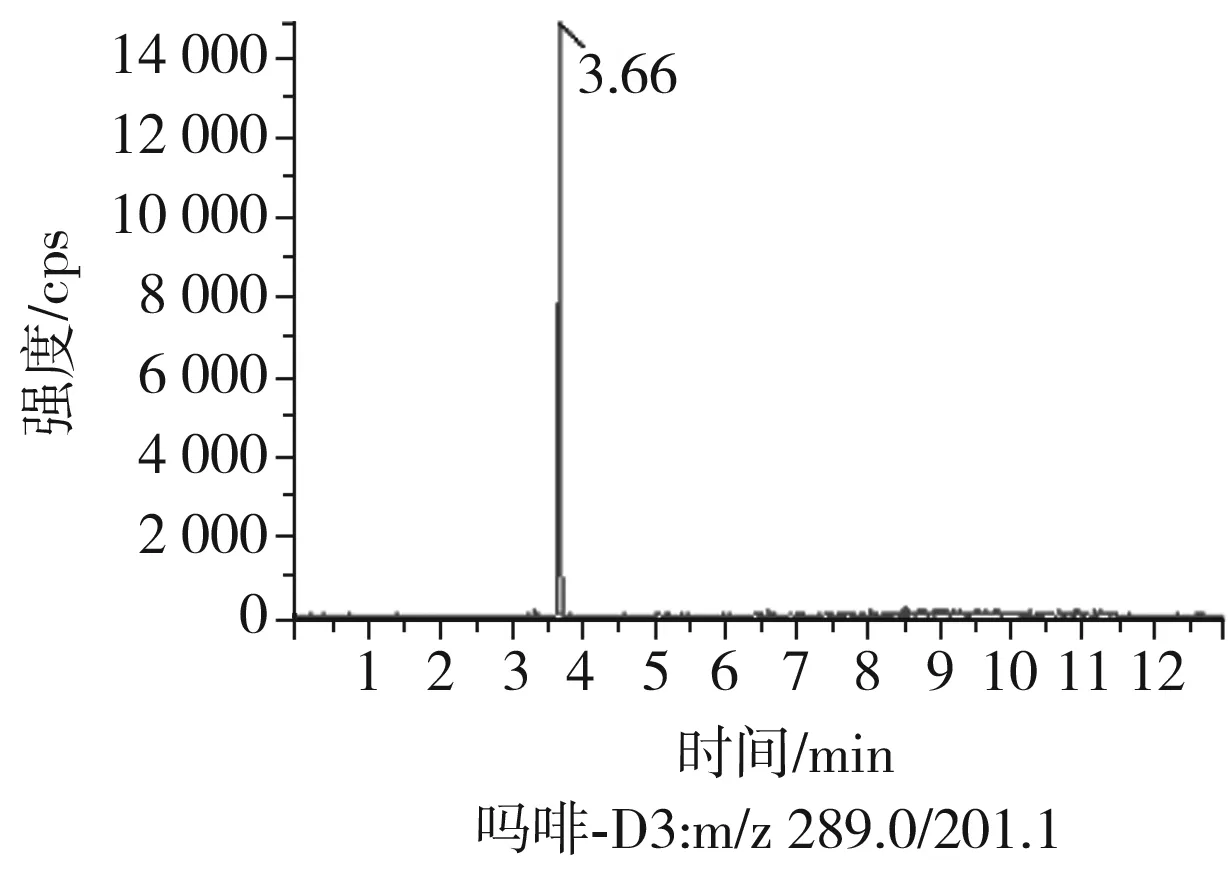
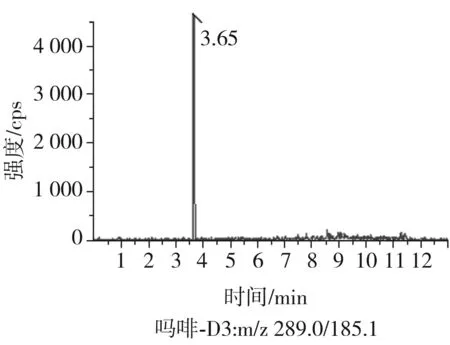
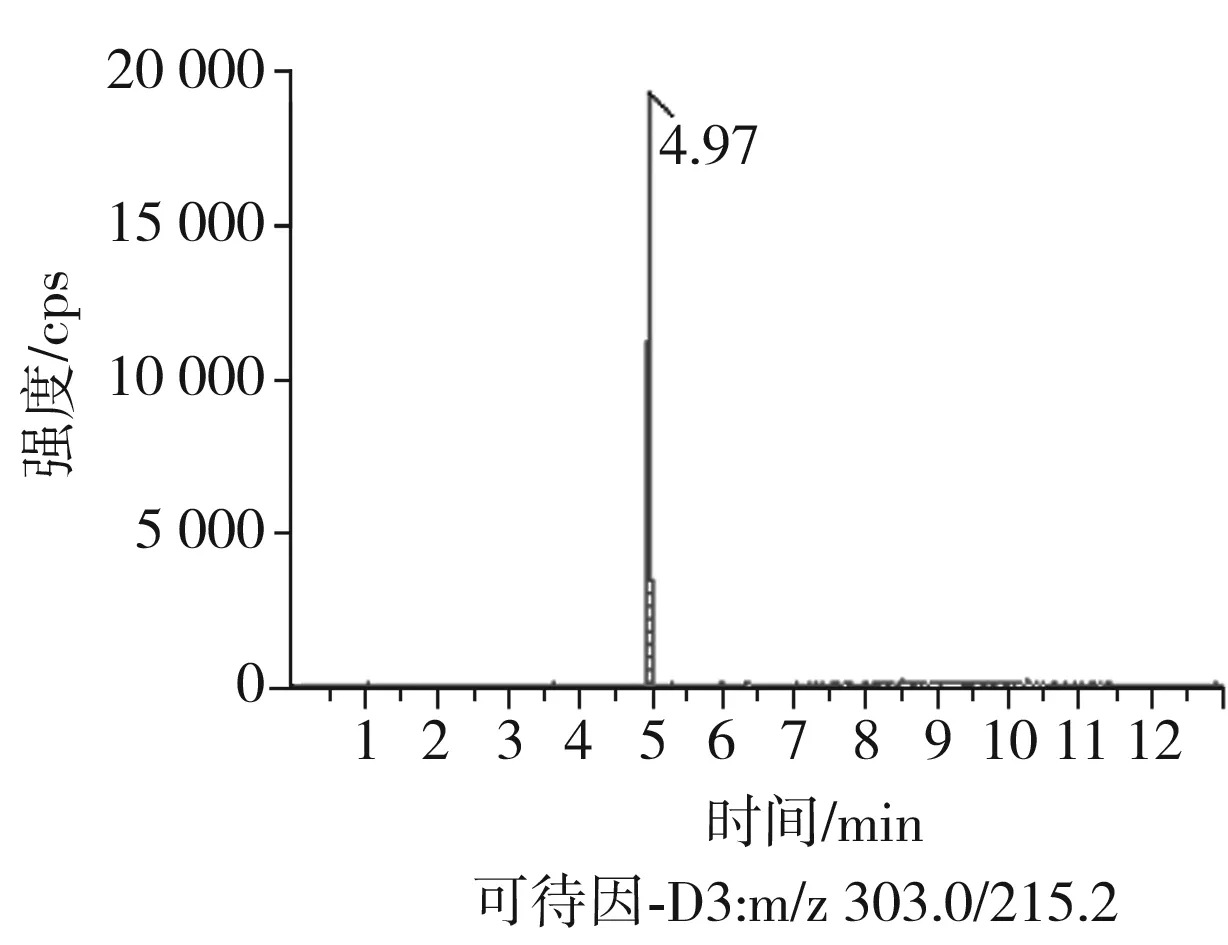
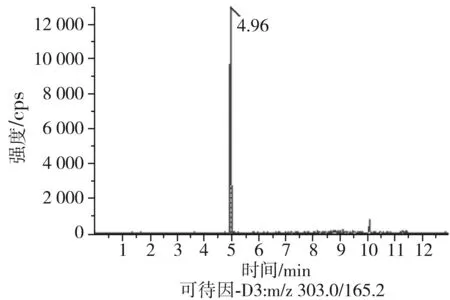
圖4 5種生物堿混合標準工作溶液的多反應監(jiān)測(MRM)色譜圖Fig.4 Multi-reaction monitoring (MRM) chromatograms of mixed standard working solution of five kinds of alkaloids
2.4 前處理方法優(yōu)化
文獻中,樣品提取后主要有兩種凈化方法:一是QuEChERS試劑凈化,二是陽離子交換固相萃取凈化。QuEChERS凈化法操作簡單,但回收率低,而陽離子交換固相萃取凈化法操作較繁瑣,且容易堵塞小柱。本文在BJS 201802《食品中嗎啡、可待因、罌粟堿、那可丁和蒂巴因的測定》方法的基礎上進行了改進,固體樣品加3 mL水,半固體樣品加2 mL水,液體樣品不加水,加乙腈15 mL超聲提取,保證樣液中乙腈含量大于80%,可有效沉淀蛋白質(zhì),加入無水醋酸鈉促進生物堿從水相轉(zhuǎn)移至有機相,并用無水硫酸鎂去除水相,在4 ℃下9 500 r/min高速離心,有效分層去除蛋白質(zhì)、油脂等雜質(zhì)。吸取1 mL乙腈提取液氮吹近干,加1 mL 10%乙腈溶液(含0.1%甲酸)溶解殘渣,進行溶劑轉(zhuǎn)換,再在4 ℃下9 500 r/min高速離心、過膜,目的是進一步去除有機物雜質(zhì)來凈化樣液。同時,用10%乙腈溶液(含0.1%甲酸)溶解樣液,可以避免出現(xiàn)溶劑效應而導致嗎啡、可待因峰形展寬,響應值降低的情況發(fā)生。結果表明,5種生物堿的回收率均達75%以上。
2.5 方法的校準曲線和檢出限
按本文優(yōu)化的色譜質(zhì)譜條件,對混合標準工作溶液系列進行測定,以嗎啡、可待因的峰面積與相應內(nèi)標物峰面積的比值為縱坐標,嗎啡、可待因的濃度與相應內(nèi)標物濃度的比值為橫坐標,繪制內(nèi)標-標準工作曲線;以蒂巴因、那可丁、罌粟堿的峰面積為縱坐標,蒂巴因、那可丁、罌粟堿的濃度為橫坐標,繪制外標-標準工作曲線,回歸方程和相關系數(shù)見表3。結果表明,嗎啡、可待因在0.50~20.0 ng/mL,蒂巴因在0.10~4.0 ng/mL,那可丁、罌粟堿在0.05~2.0 ng/mL范圍內(nèi)均有良好的線性關系,相關系數(shù)r均大于0.998。另外,在半固態(tài)調(diào)味醬空白樣品中加入低濃度混合標準工作液,制備加標樣品,按本文方法進行提取測定并計算其信噪比,以3倍信噪比(S/N=3)對應的加標量為最低檢出限。由嗎啡、可待因、蒂巴因、那可丁、罌粟堿的信噪比圖(見圖5~圖9)可知,嗎啡、可待因的方法檢出限為1.0 μg/kg,蒂巴因的方法檢出限為0.2 μg/kg,那可丁、罌粟堿的方法檢出限為0.1 μg/kg。該方法檢出限低,適用于調(diào)味料中嗎啡、可待因、蒂巴因、那可丁和罌粟堿的痕量分析。
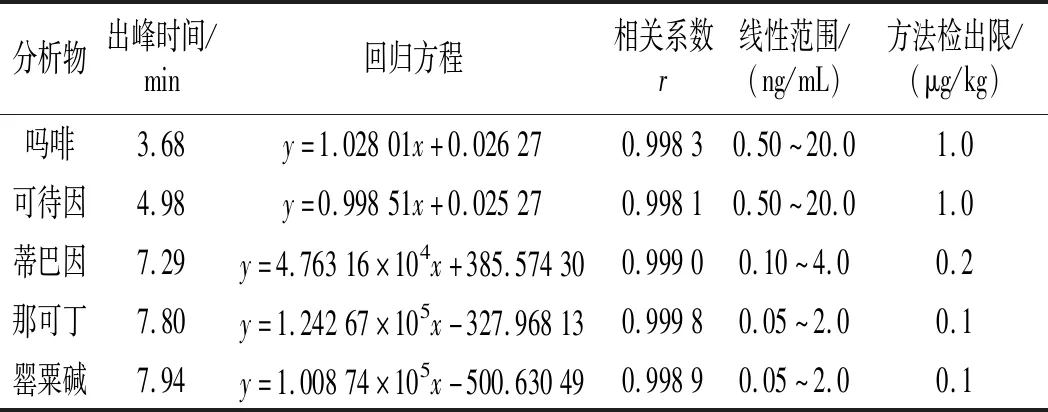
表3 標準曲線的回歸分析和方法檢出限Table 3 Regression analysis and method detection limits of standard curves
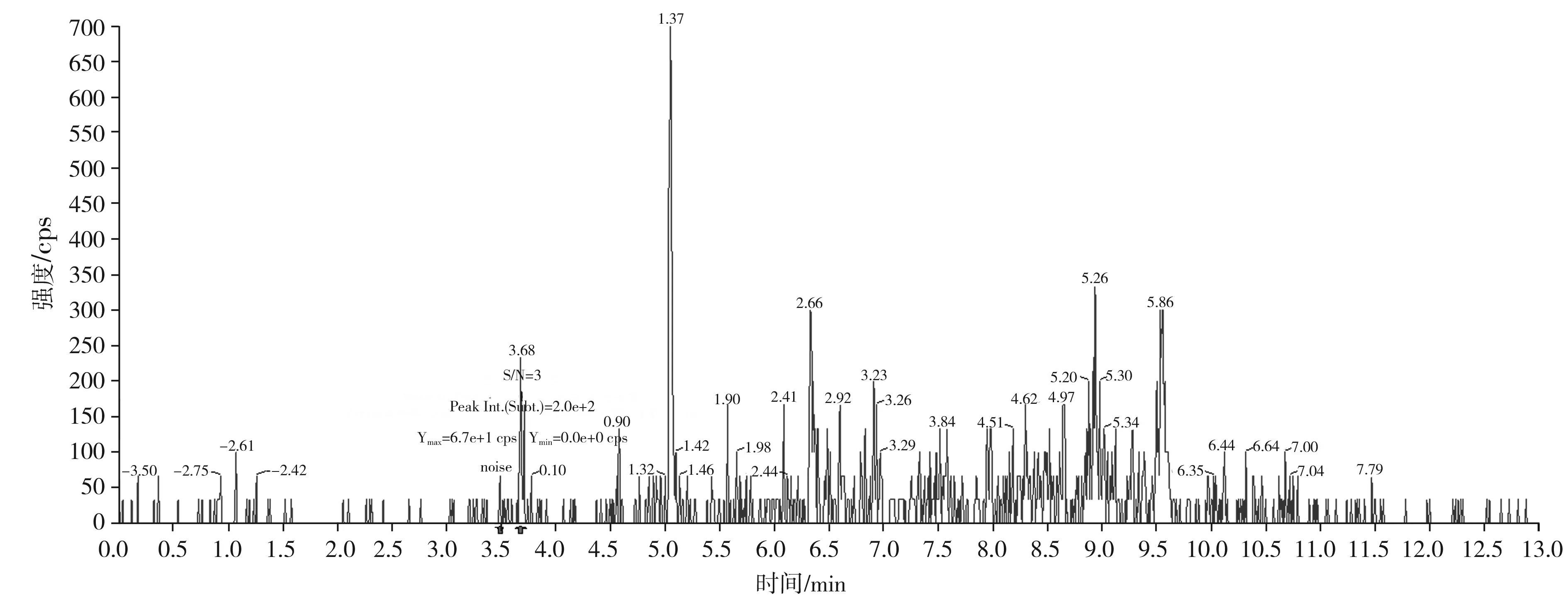
圖5 嗎啡定量離子對286.0/165.1的信噪比圖(加標量1.0 μg/kg)Fig.5 The signal to noise ratio diagram of morphine quantitative ion pair 286.0/165.1 (with adding standard matter amount of 1.0 μg/kg)
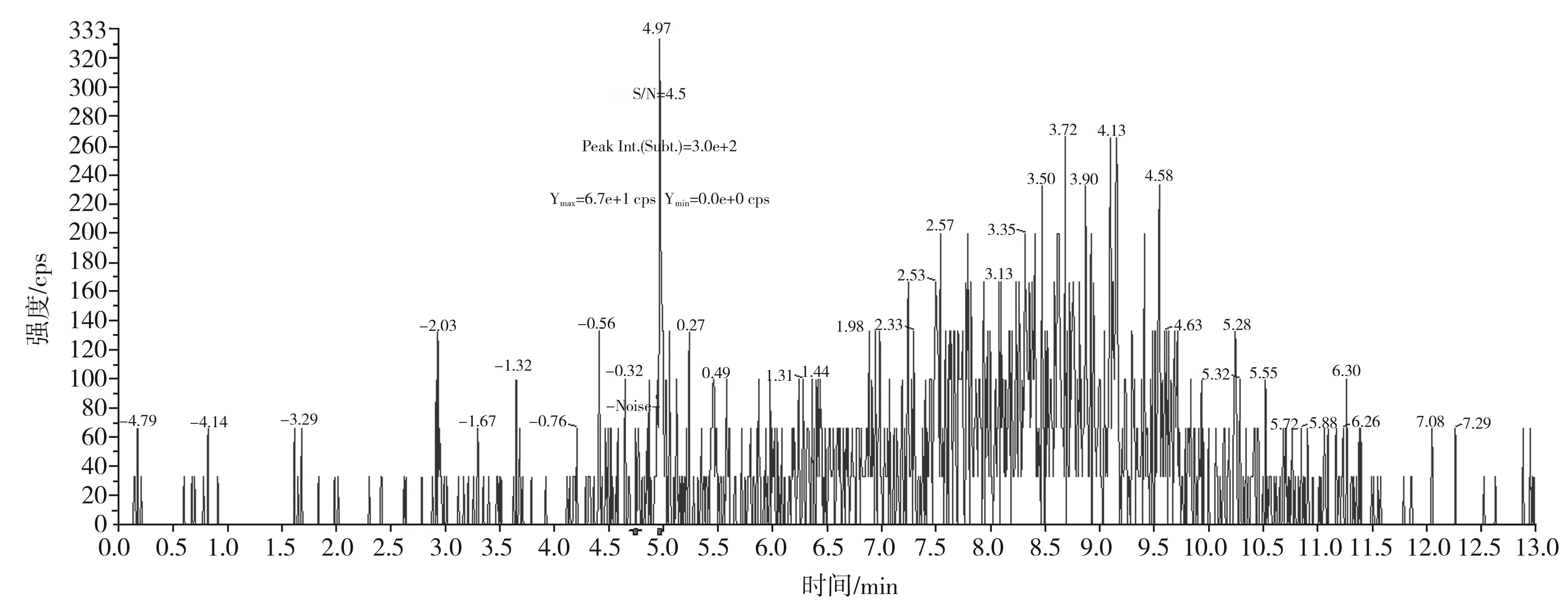
圖6 可待因定量離子對300.0/215.1的信噪比圖(加標量1.0 μg/kg)Fig.6 The signal to noise ratio diagram of codeine quantitative ion pair 300.0/215.1(with adding standard matter amount of 1.0 μg/kg)
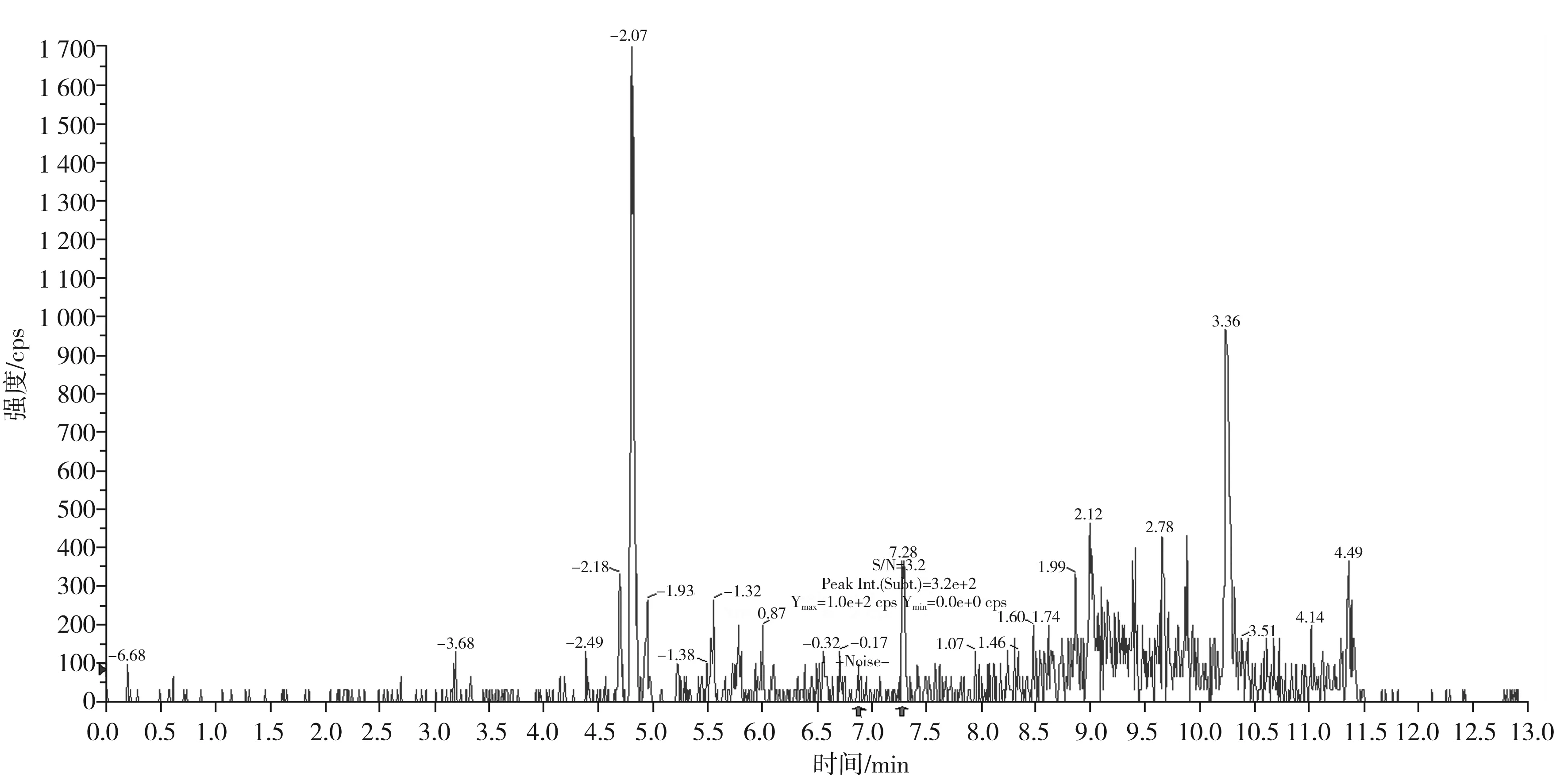
圖7 蒂巴因定量離子對312.0/58.2的信噪比圖(加標量0.2 μg/kg)Fig.7 The signal to noise ratio diagram of thebaine quantitative ion pair 312.0/58.2(with adding standard matter amount of 0.2 μg/kg)
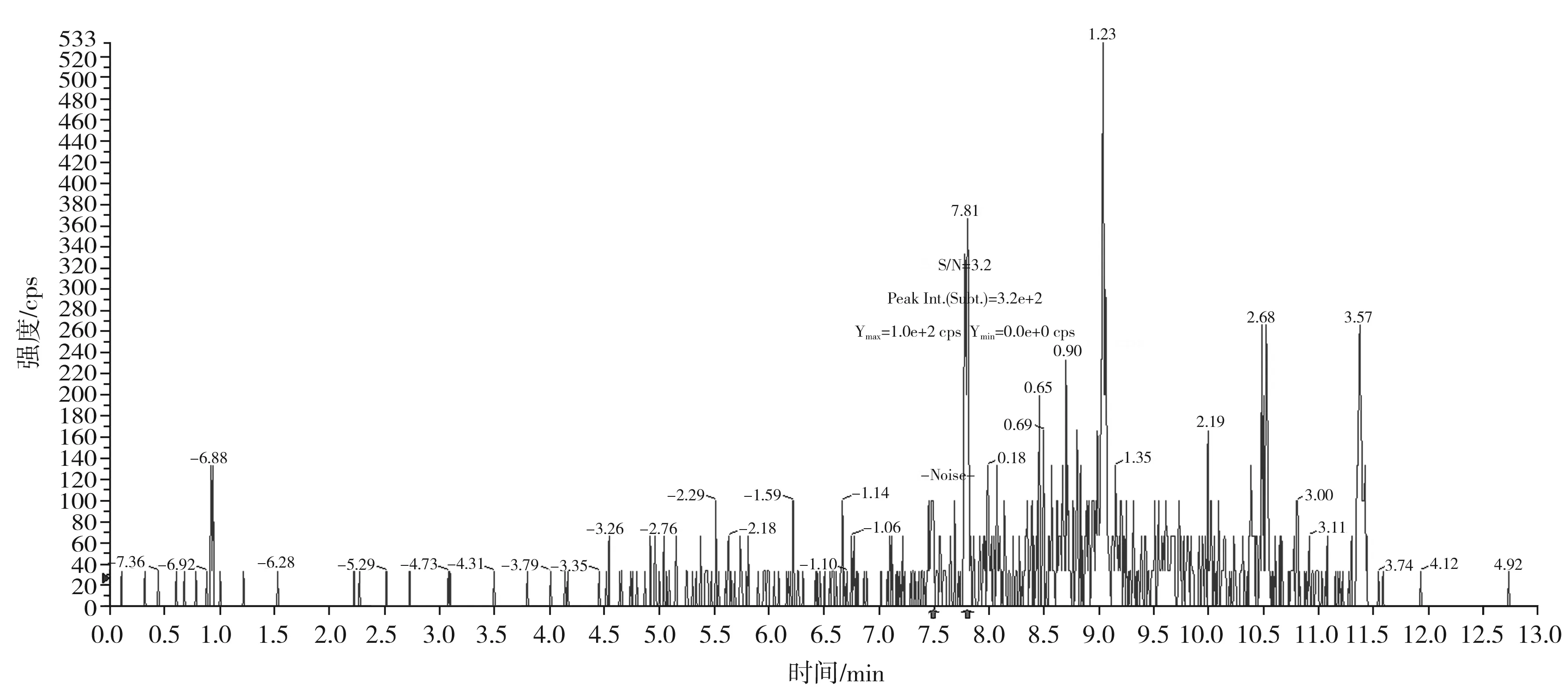
圖8 那可丁定量離子對414.2/220.2的信噪比圖(加標量0.1 μg/kg)Fig.8 The signal to noise ratio diagram of narcotine quantitative ion pair 414.2/220.2(with adding standard matter amount of 0.1 μg/kg)
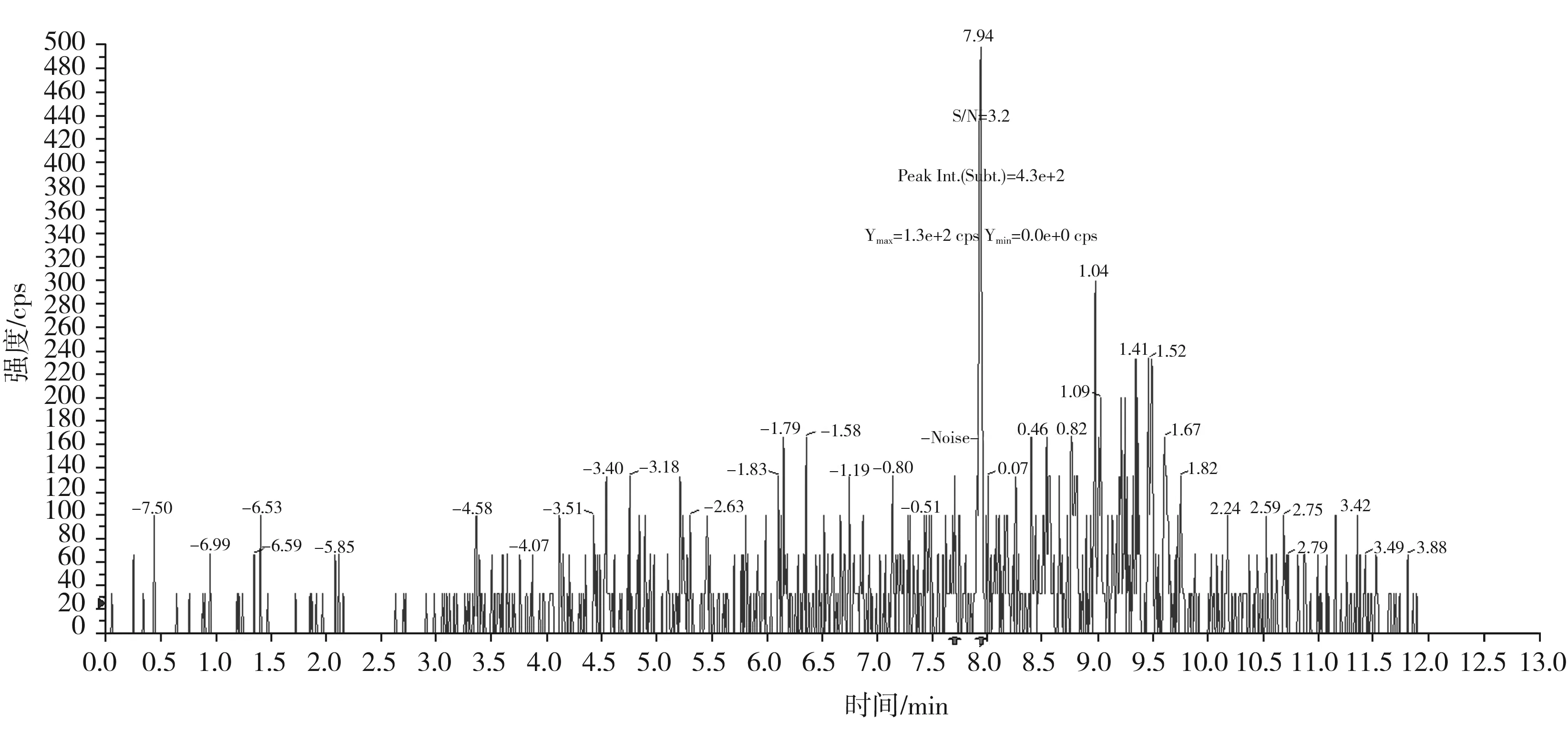
圖9 罌粟堿定量離子對340.2/202.1的信噪比圖(加標量0.1 μg/kg)Fig.9 The signal to noise ratio diagram of papaverine quantitative ion pair 340.2/202.1(with adding standard matter amount of 0.1 μg/kg)
2.6 方法的精密度和加標回收率
分別選取固態(tài)、半固態(tài)、液態(tài)調(diào)味料為空白樣品(檢測結果均為陰性),分別按5,10,20倍檢出限3個濃度水平加入5種生物堿混合標準使用液,制備加標樣品,對每個添加水平重復測定6次,計算加標回收率和相對標準偏差,數(shù)據(jù)見表4。結果表明,嗎啡回收率在76.6%~117.3%范圍內(nèi),相對標準偏差在11.9%~14.4%之間;可待因的回收率在84.4%~111.4%范圍內(nèi),相對標準偏差在4.2%~14.1%之間;蒂巴因的回收率在85.1%~111.5%范圍內(nèi),相對標準偏差在2.7%~7.1%之間;那可丁的回收率在75.2%~103.6%范圍內(nèi),相對標準偏差在2.6%~7.2%之間;罌粟堿的回收率在83.0%~106.0%范圍內(nèi),相對標準偏差在1.9%~5.3%之間,滿足GB/T 27404-2008《實驗室質(zhì)量控制規(guī)范 食品理化檢測》中附錄F對精密度、回收率的要求,說明該方法準確、穩(wěn)定、可靠。
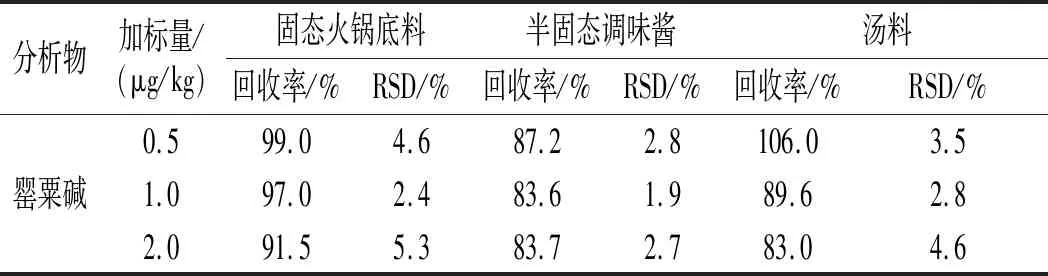
續(xù) 表
2.7 樣品測定
實驗共選取10種調(diào)味料進行了檢測,其中固態(tài)火鍋底料、半固態(tài)米線調(diào)味醬、半固態(tài)小龍蝦調(diào)味醬、湯料共4種預包裝調(diào)味料購于超市;海鮮汁、沙茶醬、孜然油、麻油、辣椒油、芝麻油共6種自制火鍋調(diào)味料(蘸料)購于火鍋店,均未檢測出嗎啡、可待因、蒂巴因、那可丁、罌粟堿,說明目前該類市售調(diào)味料質(zhì)量狀況良好,結果令人滿意。
3 結論
本試驗采用實驗室常用的Kinetex?2.6 μm Biphenyl 100 ?反相色譜柱,建立了超高效液相色譜-串聯(lián)質(zhì)譜法測定調(diào)味料中罌粟殼類生物堿,嗎啡等5種生物堿均得到了很好的色譜保留和分離,降低了方法檢出限;在樣品處理上進行了改進,獲得了理想的凈化、回收效果,使樣品前處理過程更加簡便。該方法檢出限在0.1~1.0 μg/kg之間,加標回收率在75.2%~117.3%范圍內(nèi),相對標準偏差均小于15%,具有較高的靈敏度、準確度和重現(xiàn)性,可用于調(diào)味料中嗎啡、可待因、蒂巴因、那可丁和罌粟堿的痕量分析,且該方法更容易推廣應用。
