擬輪枝鐮孢與玉米籽粒互作的差異基因篩選及代謝通路分析
2023-04-11 03:12:00渠清劉寧鄒金鵬張雅璇賈慧孫蔓莉曹志艷董金皋
中國農業科學 2023年6期
關鍵詞:植物
渠清,劉寧,鄒金鵬,張雅璇,賈慧,孫蔓莉,曹志艷,董金皋
擬輪枝鐮孢與玉米籽粒互作的差異基因篩選及代謝通路分析

1河北農業大學生命科學學院,河北保定 071001;2河北省植物生理與分子病理學重點實驗室/華北作物改良與調控國家重點實驗室,河北保定 071001;3河北農業大學植物保護學院,河北保定 071001
【目的】擬輪枝鐮孢()引起的玉米穗腐病是我國玉米產區發生嚴重的病害之一,論文旨在了解病原菌與玉米籽粒互作過程中的基因表達差異,為揭示病原菌的致病機制和玉米抗病機制提供依據。【方法】對擬輪枝鐮孢侵染玉米籽粒0、4、12和72 h的樣品進行轉錄組測序,之后采用生物信息學分析,分別以玉米和擬輪枝鐮孢基因組為參考,以|log2FC|≥1,-adjust<0.05為閾值篩選互作過程中玉米和擬輪枝鐮孢的差異表達基因,利用GO和KEGG對其進行功能注釋及富集分析。Goatools軟件分析植物-病原互作、MAPK途徑和植物激素信號轉導通路相關差異基因的表達變化,采用實時熒光定量PCR(qRT-PCR)方法對測序差異基因進行驗證。【結果】在互作4、12和72 h后擬輪枝鐮孢分別有140、400和1 945個基因上調表達,有9、302和1 784個基因下調表達;玉米分別有293、692 和1 426個基因上調表達,320、482和153個基因下調表達。GO和KEGG富集分析顯示,侵染早期擬輪枝鐮孢在細胞間隙生長,差異基因主要富集在RNA生物合成、細胞壁結構成分、脂肪酸生物合成、蛋白質代謝、碳水化合物代謝、生物過程和代謝過程等通路中。擬輪枝鐮孢早期侵染觸發了玉米活性氧(ROS)爆發,差異基因主要富集在對活性氧、過氧化氫的反應,幾丁質酶、單加氧酶活性,木質素代謝過程等相關通路中。侵染后期擬輪枝鐮孢繼續在籽粒中定殖及擴展,差異基因主要富集在碳水化合物和細胞壁多糖分解代謝過程、跨膜轉運、氧化還原酶活性等功能通路中。玉米主要通過苯丙素、木質素、類黃酮生物合成,MAPK 信號通路、植物-病原互作和植物激素信號轉導等途徑差異基因大量表達響應病原菌侵染。隨機選取6個玉米和6個擬輪枝鐮孢的差異表達基因進行qRT-PCR分析,基因表達規律與轉錄組測序結果一致,證實了RNA-seq的準確性。【結論】在病原菌侵染早期,擬輪枝鐮孢在細胞間隙生長,觸發玉米活性氧爆發,相關通路差異基因表達;侵染中后期,病原菌以淀粉為營養素,繼續在籽粒中定殖及擴展,玉米通過苯丙素、木質素及幾丁質酶的生物合成等方面的相關基因表達響應擬輪枝鐮孢侵染,同時植物-病原互作、MAPK途徑和激素信號轉導等途徑參與抗擬輪枝鐮孢侵染。
擬輪枝鐮孢;玉米穗腐病;轉錄組;植物-病原互作;基因表達; 差異表達基因
0 引言
【研究意義】玉米穗腐病是由包括鐮孢菌和霉菌在內的多種病原菌侵染玉米果穗引起的重要病害,在我國多個玉米產區均有發生。其中擬輪枝鐮孢()是玉米穗腐病的主要致病菌之一,在我國多個地區玉米穗腐病致病真菌中所占比例最大[1]。該病害除了降低玉米產量外,擬輪枝鐮孢還產生有毒的次級代謝產物——伏馬毒素,嚴重影響玉米質量。伏馬毒素可分為伏馬毒素B1(FB1)、B2(FB2)和B3(FB3)3種,其中FB1含量最多且毒性最強[2-3],其不僅可毒害動物,造成馬白頭軟化癥、豬肺水腫和大鼠肝癌,還與人類癌癥有關[4-5],嚴重影響食品及飼料安全。目前對玉米穗腐病的防治主要以種植抗病品種為主,但玉米與病原菌互作過程中植物抗病應答機制和病原菌的致病機制仍不清楚,有待于進一步深入解析其相互作用機理,為抗病分子育種提供參考。【前人研究進展】植物與病原真菌互作過程中,真菌可通過侵染墊、附著胞、吸器等侵染結構幫助其成功入侵寄主,造成植物感染[6]。鐮孢菌沒有穿透宿主細胞的特殊結構,如附著胞或吸器,但其孢子可通過植物的自然孔口或者傷口進入宿主細胞完成侵入[7]。此外,還可產生一系列細胞壁降解酶,不僅能夠消化植物細胞壁的纖維素、木聚糖和果膠的聚合物并以此作為真菌的重要營養來源,還能通過降解植物蠟質層、角質層和細胞壁而導致宿主發病[8],從而完成入侵和病原體傳播。在互作過程中,真菌還會產生毒素。真菌釋放堿性蛋白導致酶活性發生變化,受到伏馬毒素FB1污染的谷物種子表現出胚乳降解、淀粉顆粒周圍缺少蛋白質基質的性狀,這種毒素通過積累有毒鞘脂中間產物破壞植物和動物的質膜[9]。這些中間產物破壞了鞘脂的從頭合成,抑制了神經酰胺合成酶[10],因此,破壞了細胞信號和功能,改變了細胞凋亡和復制。植物擁有復雜的免疫系統,可以保護其免受病原菌的侵害,包括病原體相關分子模式(pathogen-associated molecular pattern,PAMP)觸發免疫(pattern-triggered immunity,PTI)和效應器觸發免疫(effector-triggered immunity,ETI)[11]。PTI和ETI觸發信號級聯,包括細胞內Ca2+濃度的改變、活性氧(reactive oxygen species,ROS)及次級信號分子的產生等。鈣結合蛋白可以檢測到細胞內Ca2+水平的增加,作為第二信使調控下游基因表達,在植物抗病中發揮重要作用。同時ROS生成速率的快速增加也是植物應對病原體侵染的基本反應,稱為“氧化爆發”[12]。ROS能夠激活絲裂原活化蛋白激酶(MAPK)級聯途徑,上調特定應激相關基因的表達[13]。植物激素在調節植物對各種生物和非生物脅迫的反應所涉及的發育過程和信號網絡中同樣發揮著重要作用[14]。大量研究顯示,植物產生的乙烯(ET)、水楊酸(SA)、茉莉酸(JA)在介導植物抗病反應中起主要作用。當病原菌侵染時,這些植物激素的合成相關基因表達水平增加,又進一步誘導和加強了植物免疫反應,實現對不同病害的抵抗[15-17]。【本研究切入點】本課題組前期對我國多個省份玉米果穗的穗腐病病原菌進行分離,發現擬輪枝鐮孢的分離頻率最高,同時通過熒光標記菌株研究多種鐮孢菌侵染玉米果穗的效能,發現擬輪枝鐮孢在侵染玉米果穗中表現出競爭優勢[18]。目前對擬輪枝鐮孢侵染玉米籽粒過程中,不同抗/感玉米品種響應病原菌侵染相關過程的轉錄分析較多[19-20],但對宿主與病原菌雙方互作過程的進程開展系統的轉錄水平研究較少。【擬解決的關鍵問題】分別以單獨培養的擬輪枝鐮孢和未接菌的玉米籽粒為對照,利用高通量轉錄測序技術對擬輪枝鐮孢侵染玉米籽粒4、12和72 h的樣品進行轉錄組測序,分析互作過程中玉米和擬輪枝鐮孢雙方的差異基因表達情況,明確擬輪枝鐮孢與玉米互作的生物學過程,以期為揭示寄主和病原菌互作及穗腐病抗性機理提供參考依據。
1 材料與方法
試驗于2020—2022年在河北農業大學完成。
1.1 供試材料
供試植物:玉米品種為自交系B73,種子由沈陽農業科學院劉新芳老師饋贈。玉米B73種植于河北農業大學東校區溫室,行間距為60 cm,株間距為30 cm,正常管理。
供試病原菌:供試病原菌為擬輪枝鐮孢Fv7600,菌株由南京農業大學顧沁老師饋贈,河北農業大學真菌毒素與植物分子病理學實驗室保存。接種于PDA(potato dextrose agar)培養基,于25℃黑暗培養。
1.2 病原菌接種
將PDA平板上活化后的擬輪枝鐮孢用直徑為6 mm的打孔器打取菌盤,接種到羧甲基纖維素鈉(CMC)液體培養基中25℃黑暗振蕩培養5—7 d后,三層無菌紗布過濾,無菌水稀釋成1×106個/mL的孢子懸浮液備用。待自交系玉米B73人工授粉21 d后,用1 mL注射器針頭蘸取擬輪枝鐮孢孢子懸浮液針刺玉米籽粒接種[21]。分別于接種后0 h(mock)、4 h(Fv4)、12 h(Fv12)和72 h(Fv72)后取樣,每個時間點接種3個果穗作為生物學重復。同時使用PDA培養基上25℃黑暗培養72 h的擬輪枝鐮孢(Fv)作為真菌的對照。所有樣品取樣后立即在液氮中冷凍,-80℃下儲存,以提取RNA。
1.3 轉錄組測序
1.3.1 RNA提取、文庫構建及測序 轉錄組測序是基于Illumina Novaseq 6000 測序平臺,對真菌菌絲、玉米籽粒和互作時期的玉米籽粒樣品轉錄出來的所有mRNA進行測序,測序試驗采用Illumina TruseqTMRNA sample prep Kit方法進行文庫構建,具體如下:使用Trizol法分別提取真菌菌絲、玉米籽粒和互作玉米樣品總RNA。用1%瓊脂糖凝膠電泳分析所提取的總RNA的完整性及是否存在DNA污染,使用NanoDrop2000(Thermo Fisher Scientific)和Agilent 2100 Nano(Fisher Scientific)儀器分析總RNA的質量和完整性。構建了15個RNA文庫(分別命名為Mock、Fv、Fv4、Fv12和Fv72,每處理3個重復)。轉錄組測序由上海美吉生物處理完成。使用Novaseq 6000測序平臺獲得原始數據,通過對原始測序數據進行質控,得到高質量的數據并對其進行統計以及質量評估,質控后得到 clean reads。將質控后的clean reads分別與玉米和擬輪枝鐮孢的參考基因組比對。使用StringTie軟件對每個樣本進行拼接組合,對拼接之后的結果進行統計。數據已上傳NCBI數據庫,SRA登錄號為PRJNA735336。
1.3.2 差異表達基因(DEG)的篩選 利用RSEM表達定量軟件分別對基因和轉錄本的表達水平進行定量分析,定量指標為TPM,即以轉錄本的條數為計算單位,使用轉錄本的條數代替拼接片段數。使用DESeq2軟件以mRNA的-adjust<0.05&|log2FC|≥1為標準對讀取clean reads進行統計分析,獲得差異表達的mRNA(DEG)。以歸一化處理后的log2FC的值繪制熱圖。
1.3.3 差異表達基因GO和KEGG富集分析 采用Goatools軟件,Fisher 精確檢驗的方法對差異表達基因進行基因本體論GO富集分析以及京都基因和基因組百科全書KEGG富集分析,從而獲得差異基因主要GO功能和KEGG富集通路。當經過校正的值(value)<0.05時,認為此GO功能和KEGG功能存在顯著富集。
1.4 實時熒光定量PCR(qRT-PCR)驗證
隨機選取6個玉米的差異表達基因和6個擬輪枝鐮孢的差異表達基因,對其進行qRT-PCR檢測。使用RNA提取試劑盒提取總RNA(北京聚合美生物科技有限公司),采用cDNA PrimeScriptTMRT reagent Kit with gDNA Eraser第一鏈合成試劑盒(寶生物工程(大連)有限公司)反轉錄合成cDNA,將其作為模板,使用Super EvaGreen qPCR Master Mix 試劑盒(US EVERBRIGHT INC 公司)進行qRT-PCR檢測,玉米使用作為內參基因,擬輪枝鐮孢使用作為內參基因,引物由生工生物工程(上海)股份有限公司合成(表1)。20 μL qPCR反應體系:SYBR Mix 10 μL、正反向引物各0.4 μL、cDNA模板1 μL、ROX 1 μL和ddH2O 7.2 μL。反應程序:95℃預變性 2 min;95℃變性5 s,60℃退火30 s,45個循環。試驗數據采用GraphPad Prism 8軟件進行統計分析,應用檢驗法進行差異顯著性檢驗。
2 結果
2.1 轉錄組測序結果評估
對接種擬輪枝鐮孢后0、4、12和72 h的玉米籽粒樣品和培養基上培養3 d的擬輪枝鐮孢進行轉錄組測序。本研究構建了15個轉錄組文庫,其中包括5個樣本,每個樣本有3個生物學重復。15個轉錄組文庫的測序結果顯示,在去除reads中的接頭序列、低質量片段和N率(N表示不確定堿基信息)較高序列及長度過短序列后,總共獲得1 169 261 200 clean reads。接菌后0 h的樣品(Mock)分別獲得了51 110 882、50 311 356和44 387 490 clean reads。真菌菌絲樣品(Fv)的3個重復數據顯示,分別獲得了45 922 668、49 966 126和40 694 544 clean reads。接菌后4 h的樣品(Fv4)獲得了101 474 514、95 247 160和83 492 592 clean reads。接菌后12 h的樣品(Fv12)獲得了102 019 788、106 427 588和106 395 294 clean reads。接菌后72 h的樣品(Fv72)獲得了107 478 816、103 103 634和81 228 748 clean reads(表2)。將各樣品的clean reads分別比對到玉米B73和擬輪枝鐮孢的基因組上,玉米中的比對率介于91.63%—95.46%。由于接菌時間較短,大部分為玉米籽粒的樣品,籽粒中擬輪枝鐮孢帶菌量和所占比例相對較少,雖然互作樣品的數據比對到真菌基因組的比對率較低,但真菌的對照比對到擬輪枝鐮孢基因組的比對率均在95%以上,所有文庫的Q30均在93.59%以上。因此,測序得到的數據是可靠的,并且可以用于后續的分析。
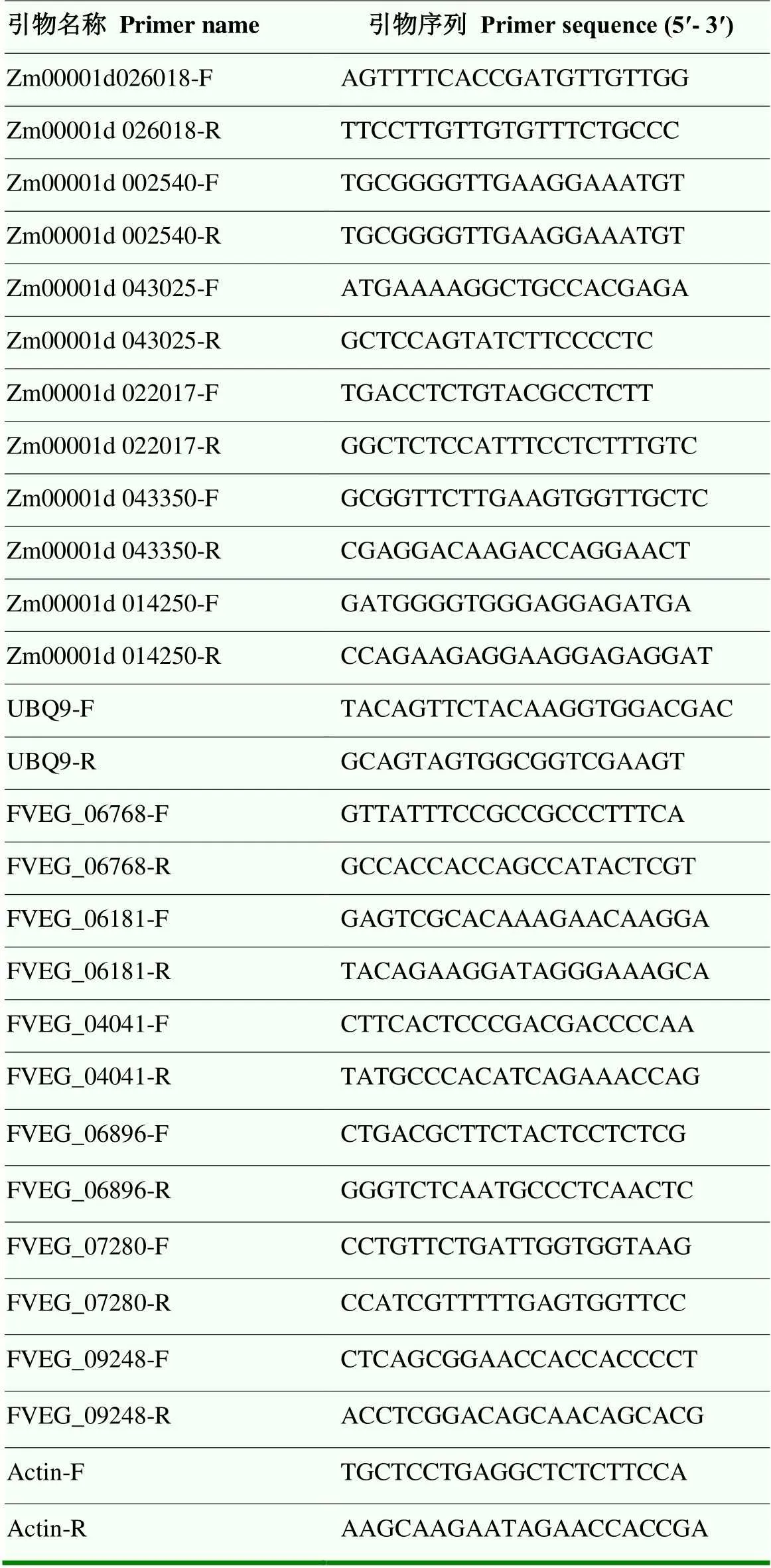
表1 本研究所用實時熒光定量 PCR引物信息
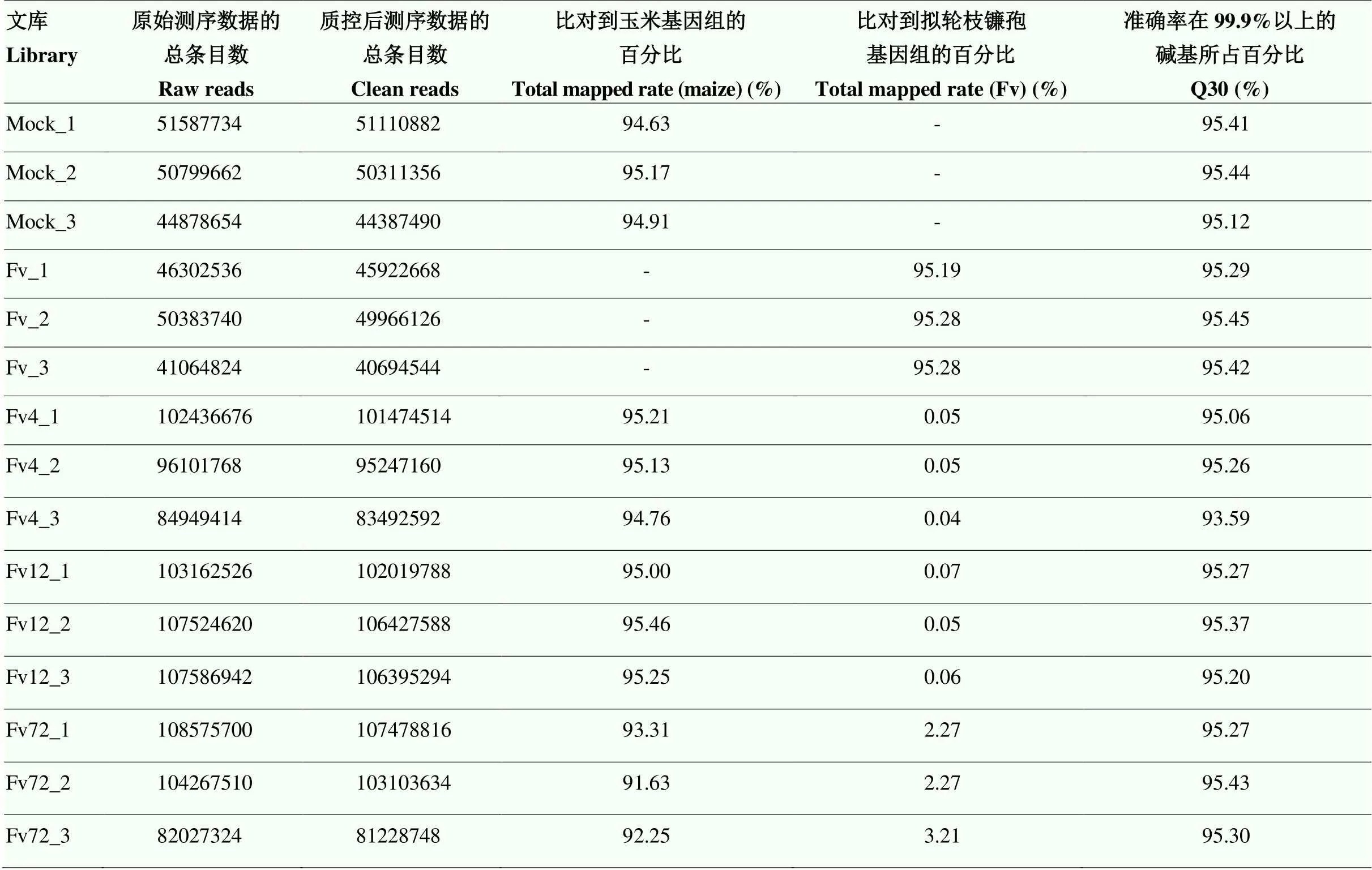
表2 轉錄組測序數據統計
-:未比對 Not blast
2.2 互作過程中擬輪枝鐮孢和玉米籽粒中差異表達的基因
2.2.1 擬輪枝鐮孢中差異表達的基因 選擇TPM>1作為基因表達的衡量標準,以PDA培養基中生長3 d的菌絲(Fv)為對照,接種擬輪枝鐮孢4 h(Fv4)、12 h(Fv12)和72 h(Fv72)的樣本計算基因表達情況,結果顯示(圖1-A),Fv中有10 618個基因表達,Fv4、Fv12和Fv72中分別有2 193、5 249和9 844個基因表達,各處理間共同表達的基因數為1 736。與Fv相比,Fv4、Fv12和Fv72中分別有5、33和422個獨特表達的基因。為了鑒定參與擬輪枝鐮孢侵染玉米籽粒的相關基因,進一步篩選了試驗組和對照組之間差異表達的基因。共篩選出4 152個差異基因,3個時間點共同差異表達的有42個基因,Fv4和Fv12 共同差異表達的有6個基因,Fv4和Fv72共同差異表達的有55個基因,Fv12和Fv72 共同差異表達的有283個基因(圖1-B)。同Fv相比,Fv4、Fv12和Fv72分別有140、400和1 945個差異基因上調表達,3個時間點共同上調表達的有38個差異基因,Fv4和Fv12共同上調表達的有4個差異基因,Fv4和Fv72共同上調表達的有47個差異基因,Fv12和Fv72共同上調表達的有151個差異基因(圖1-C)。Fv4、Fv12和Fv72分別有9、302和1 784個差異基因下調表達,3個時間點共同下調表達的有4個差異基因,Fv4和Fv12共同下調表達的有2個差異基因,Fv12和Fv72共同下調表達的有94個差異基因(圖1-D)。結果顯示,與在培養基中單獨培養相比,擬輪枝鐮孢侵染玉米籽粒的早期差異表達基因較少,菌絲以營養生長的方式在玉米籽粒中生長,但隨著時間延長,基因表達出現顯著差異,不僅激活大量基因的表達,同時抑制了相當數量的基因表達。
2.2.2 玉米中差異表達的基因 選擇TPM>1作為基因表達的衡量標準,以接種無菌水(mock)為對照,接種擬輪枝鐮孢4 h(Fv4)、12 h(Fv12)、72 h(Fv72)的樣本計算基因表達情況,結果顯示(圖2-A),對照處理中有19 706個基因表達,Fv4、Fv12和Fv72玉米籽粒中分別有20 173、20 657和20 169個基因表達,各處理間共同表達的基因數為18 743。與mock相比,Fv4、Fv12和Fv72玉米籽粒中分別有160、484和535個獨特表達的基因。為了鑒定玉米籽粒抵御擬輪枝鐮孢侵染的相關基因,進一步篩選了試驗組和對照組之間差異表達的基因。共篩選出2 778個差異基因,3個時間點共同差異表達的有64個基因,Fv4和Fv12共同差異表達的有226個基因,Fv4和Fv72共同差異表達的有69個基因,Fv12和Fv72共同差異表達的有165個基因(圖2-B)。同未接菌的玉米籽粒相比,Fv4、Fv12和Fv72分別有293、692和 1 426個差異基因上調表達,3個時間點共同上調表達的有39個差異基因,Fv4和Fv12共同上調表達的有73個差異基因,Fv4和Fv72共同上調表達的有21個差異基因,Fv12和Fv72共同上調表達的有144個差異基因(圖2-C)。Fv4、Fv12和Fv72分別有320、482和153個差異基因下調表達,3個時間點共同下調表達的有10個差異基因,Fv4和Fv12共同下調表達的有111個差異基因,Fv4和Fv72共同下調表達的有29個差異基因,Fv12和Fv72共同下調表達的有11個差異基因(圖2-D)。結果顯示,擬輪枝鐮孢侵染后玉米籽粒更多的基因顯示表達上調的趨勢,且隨著侵染時間延長上調表達基因的個數迅速增加。
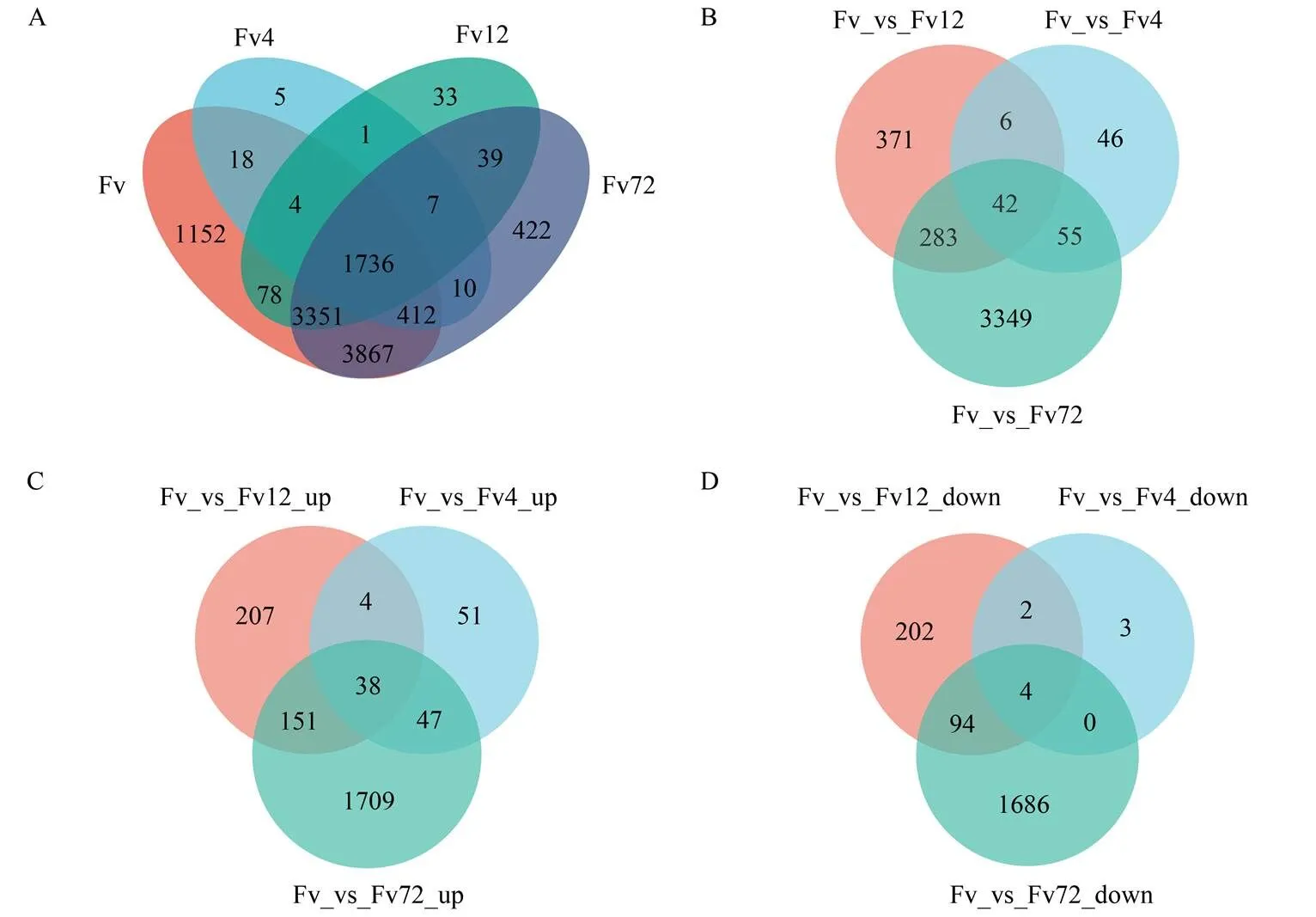
A:樣本間基因表達的維恩圖Venn of gene expression among samples;B:接種4、12和72 h擬輪枝鐮孢中差異基因的維恩圖Venn of DEGs in F. verticillioides after inoculation at 4, 12 and 72 h;C:接種4、12和72 h后擬輪枝鐮孢中上調表達差異基因的維恩圖Venn of up-regulated DEGs in F. verticillioides after inoculation at 4, 12 and 72 h;D:接種4、12 和72 h后擬輪枝鐮孢中下調表達差異基因的維恩圖Venn of down-regulated DEGs in F. verticillioides after inoculation at 4, 12 and 72 h
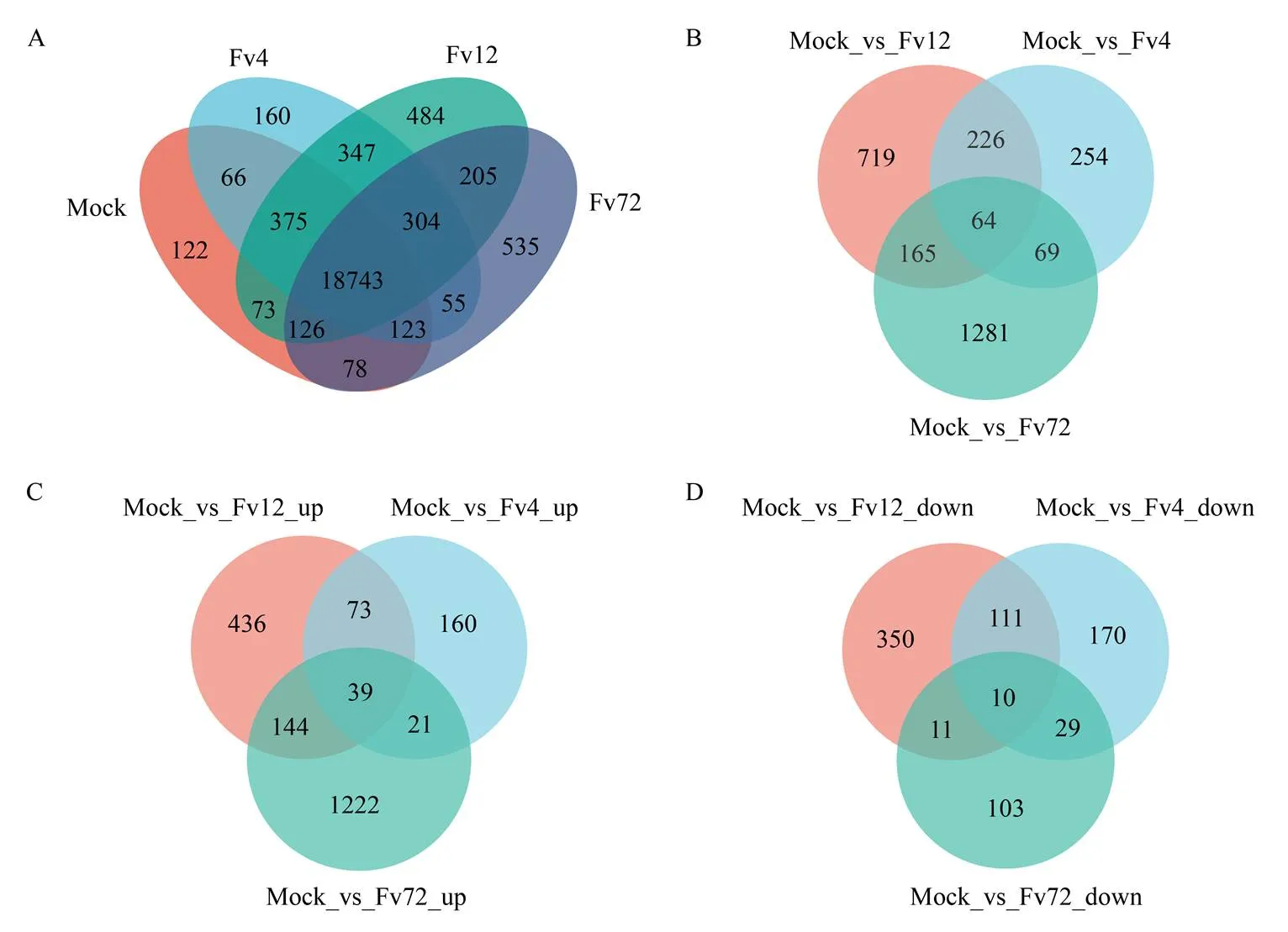
A:樣本間基因表達的維恩圖Venn of gene expression among samples;B:接種4、12和72 h玉米中差異基因的維恩圖Venn of DEGs in maize after inoculation with F. verticillioides at 4, 12 and 72 h;C:接種4、12和72 h后玉米中上調表達差異基因的維恩圖Venn of up-regulated DEGs in maize after inoculation with F. verticillioides at 4, 12 and 72 h;D:接種4、12和72 h后玉米中下調表達差異基因的維恩圖Venn of down-regulated DEGs in maize after inoculation with F. verticillioides at 4, 12 and 72 h
2.3 差異表達基因的代謝通路分析
2.3.1 擬輪枝鐮孢的差異表達基因及富集分析 對互作過程中擬輪枝鐮孢的差異基因進行了GO富集分析(表3)。侵染4 h時擬輪枝鐮孢差異基因較少,主要富集在RNA生物合成、細胞壁結構成分等功能通路中。侵染12 h后擬輪枝鐮孢差異基因主要富集在肽生物合成和代謝過程、蛋白質代謝、碳水化合物代謝、生物過程和代謝過程等功能通路中。侵染72 h后擬輪枝鐮孢的差異基因主要富集在碳水化合物和細胞壁多糖分解代謝過程、跨膜轉運、氧化還原酶活性等功能通路中。
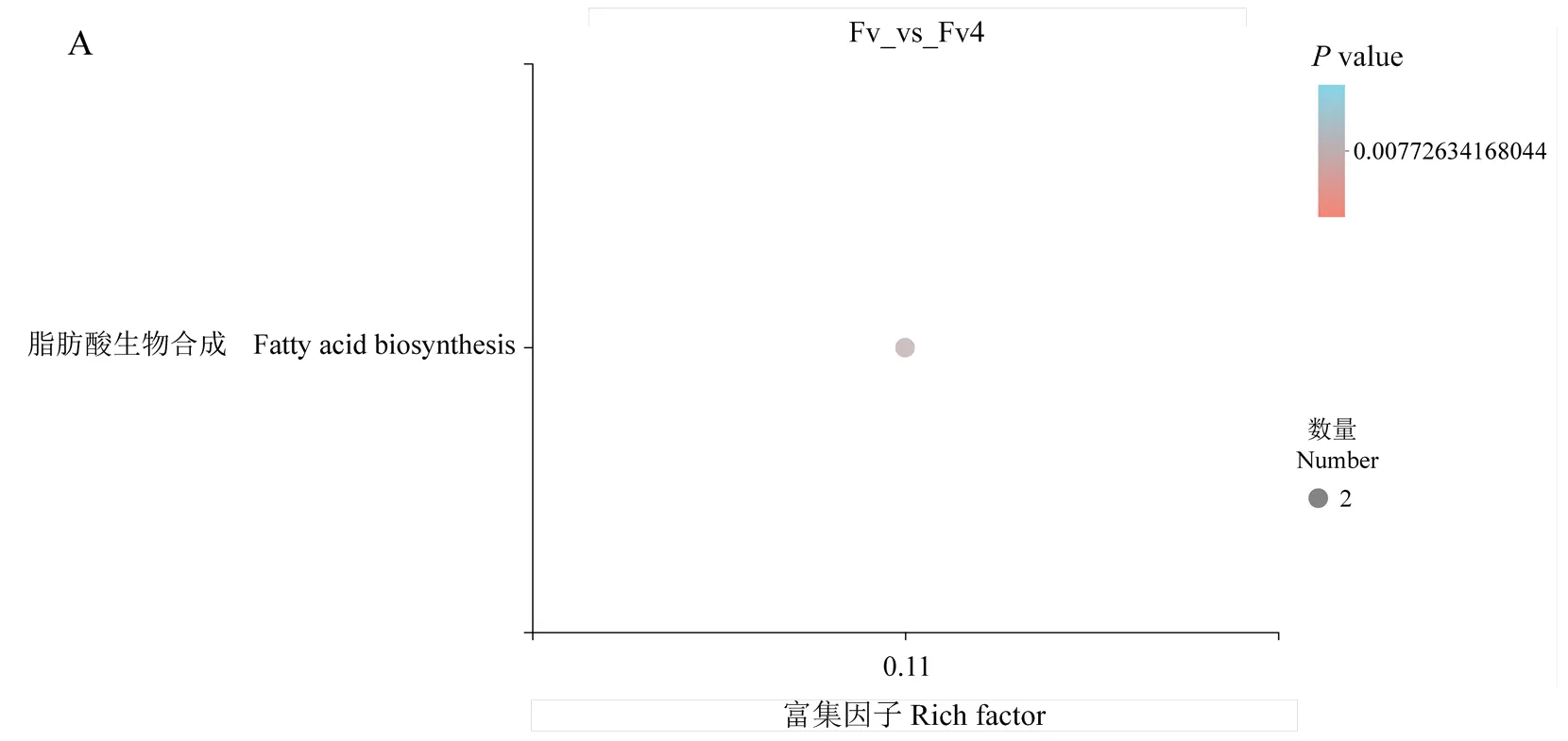
在KEGG富集分析中,侵染4 h擬輪枝鐮孢的差異基因主要富集在脂肪酸生物合成通路中(圖3-A),侵染12 h擬輪枝鐮孢的差異基因主要富集在核糖體途徑中(圖3-B),侵染72 h擬輪枝鐮孢的差異基因大量增加,主要富集在類固醇生物合成、淀粉和蔗糖代謝、戊糖和葡萄糖醛酸相互轉化及精氨酸、脯氨酸、色氨酸、甘氨酸、絲氨酸、蘇氨酸和苯丙氨酸等氨基酸的代謝通路中(圖3-C)。
2.3.2 玉米的差異表達基因及富集分析 對侵染后不同時間點的玉米差異基因進行了GO富集分析(表4)。接菌4 h后玉米的差異基因主要富集在對活性氧、過氧化氫的反應、幾丁質酶、單加氧酶活性等相關通路中。接菌12 h后玉米的差異基因主要富集在對過氧化氫、非生物刺激的反應,木質素代謝過程及幾丁質酶活性等相關通路中。病原菌侵染72 h后,差異基因主要富集在苯丙素、木質素、脂質生物合成過程,水楊酸、茉莉酸、活性氧代謝過程,幾丁質酶和苯丙氨酸解氨酶活性等相關通路中。
KEGG富集分析結果顯示,接菌后4 h玉米的差異基因主要富集在內質網中的蛋白質加工、苯丙素生物合成、MAPK信號通路中(圖4-A)。接菌后12 h玉米的差異基因主要富集在內質網中蛋白質加工、苯丙素生物合成、二苯醚類、二芳庚烷類和姜辣素生物合成、淀粉與蔗糖代謝等通路中(圖4-B)。接菌后72 h玉米的差異基因主要富集在苯丙素、二苯醚類、二芳庚烷類和姜辣素生物合成、類黃酮生物合成、MAPK 信號通路、植物-病原互作和植物信號轉導等途徑中(圖4-C)。
2.4 玉米響應擬輪枝鐮孢侵染的抗病通路
進一步分析玉米響應擬輪枝鐮孢侵染中的抗病通路,包括植物-病原互作、MAPK途徑和植物激素信號轉導3條途徑。植物-病原互作通路中,玉米響應擬輪枝鐮孢侵染的差異表達基因分別在接菌后不同時間點響應病原菌侵染(圖5)。聚類分析將該途徑的差異基因分為兩種類型,首先編碼玉米CaMCML的基因、、在接菌早期就出現上調表達,這可能是由細胞內Ca2+濃度發生快速變化引起的;而其他基因主要是在病原菌侵染12或72 h時表達開始上調,但均為72 h表達量最高。例如呼吸爆發氧化酶Rboh()在接菌12 h 就開始表達上調,72 h表達水平最高;WRKY33 轉錄因子(、)在接菌12 h后就開始上調表達,而其他WRKY33 轉錄因子(、、)在72 h表達迅速達到最大。同樣在侵染后期表達高的基因還有鈣依賴蛋白質激酶CDPK的基因(、)、誘導植物系統性抗性的基因PR1(、、)。
MAPK級聯是受體/傳感器下游高度保守的信號模塊,可將細胞外刺激轉化為真核生物的細胞內反應。植物MAPK信號通路在植物抵御病原體侵染的信號傳遞中起著關鍵作用。玉米抗擬輪枝鐮孢侵染過程中,MAPK信號通路差異基因熱圖分析發現除了個別基因()下調外,其他基因均表現為在72 h才出現表達上調(圖6),例如MAPK信號途徑中維持活性氧穩態的OXI1基因()和MPK3/6基因(),在接菌后72 h均上調表達。
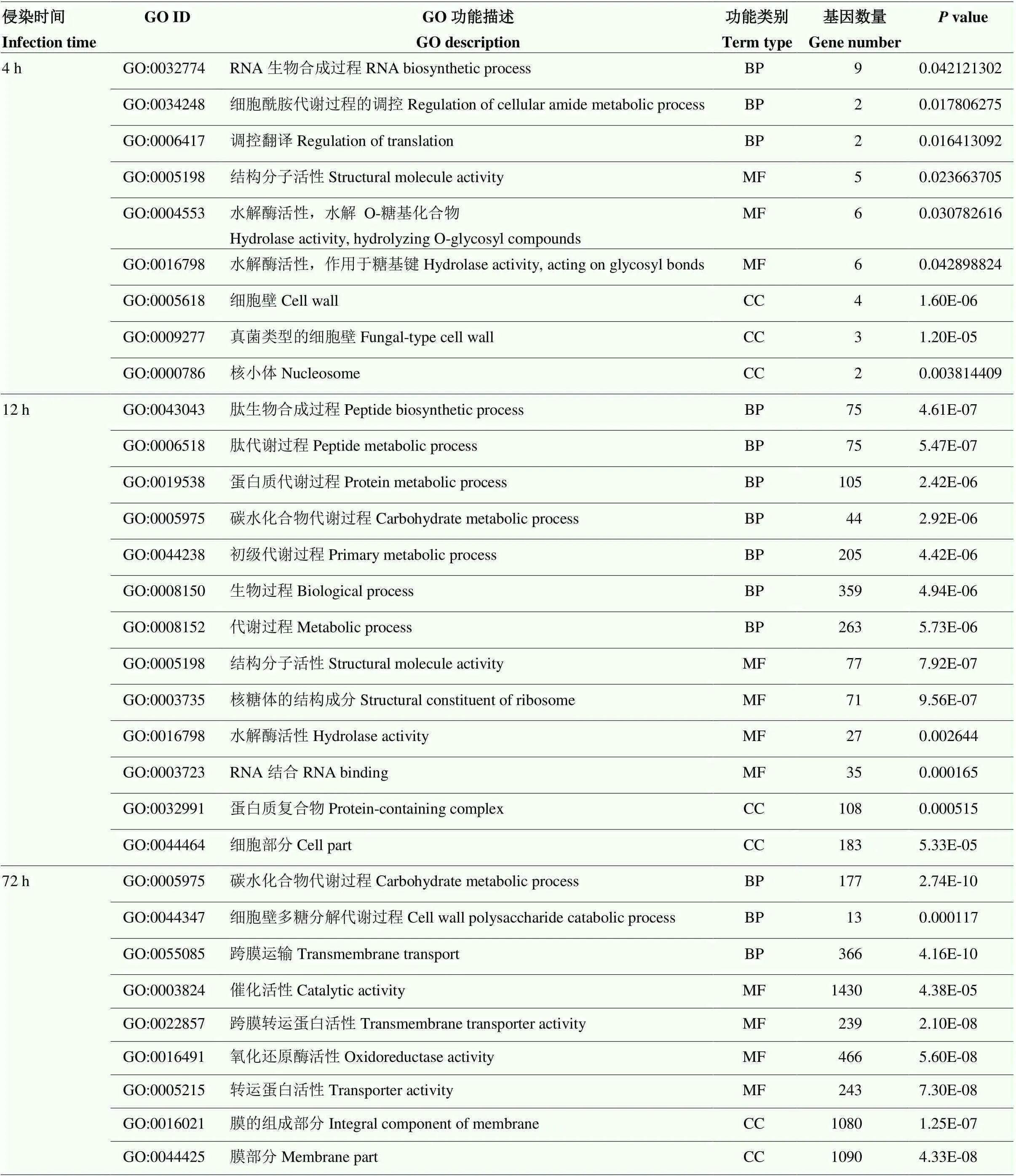
表3 擬輪枝鐮孢侵染玉米不同時間點差異表達基因的 GO富集分析
BP:生物學進程Biological process; MF:分子功能Molecular function; CC:細胞組分Cellular component。表4同The same as Table 4
植物應對各種生物和非生物脅迫中,植物激素發揮著重要作用。轉錄組結果顯示,玉米受到擬輪枝鐮孢侵染后不同的激素信號響應了病原菌的侵染,而且各通路中不同基因存在前后差異(圖7)。例如同樣是茉莉酸(JA)信號通路中的關鍵基因JAZ(),在病菌侵染12 h后就檢測到持續的顯著上調,而在接菌后72 h JAZ的另外3個基因(、、)才發生表達上調。水楊酸(SA)信號通路中參與抗病的PR1基因(、、)在接菌后72 h上調表達。同樣在后期72 h發生上調的還包括乙烯通路中EIN3基因()和ERF1/2基因(、)。這表明茉莉酸信號通路的觸發較早,而水楊酸信號通路和乙烯通路的觸發是發生在接菌后72 h。其他受影響的信號通路基因還包括油菜素內酯通路和脫落酸通路相關基因,也參與了植物-病原互作過程。
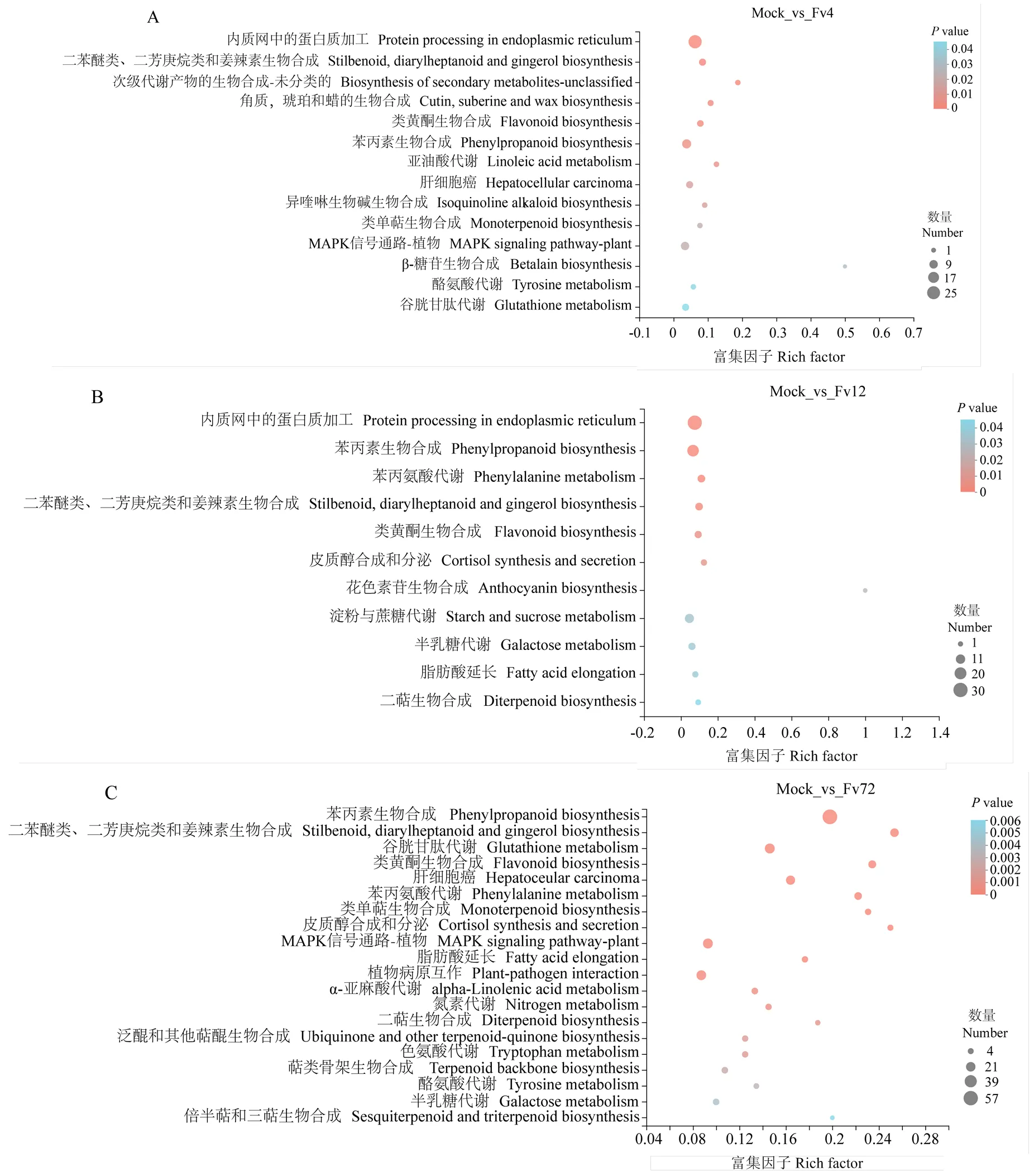
圖4 接種擬輪枝鐮孢不同時間點后玉米中差異表達基因KEGG代謝通路富集分析
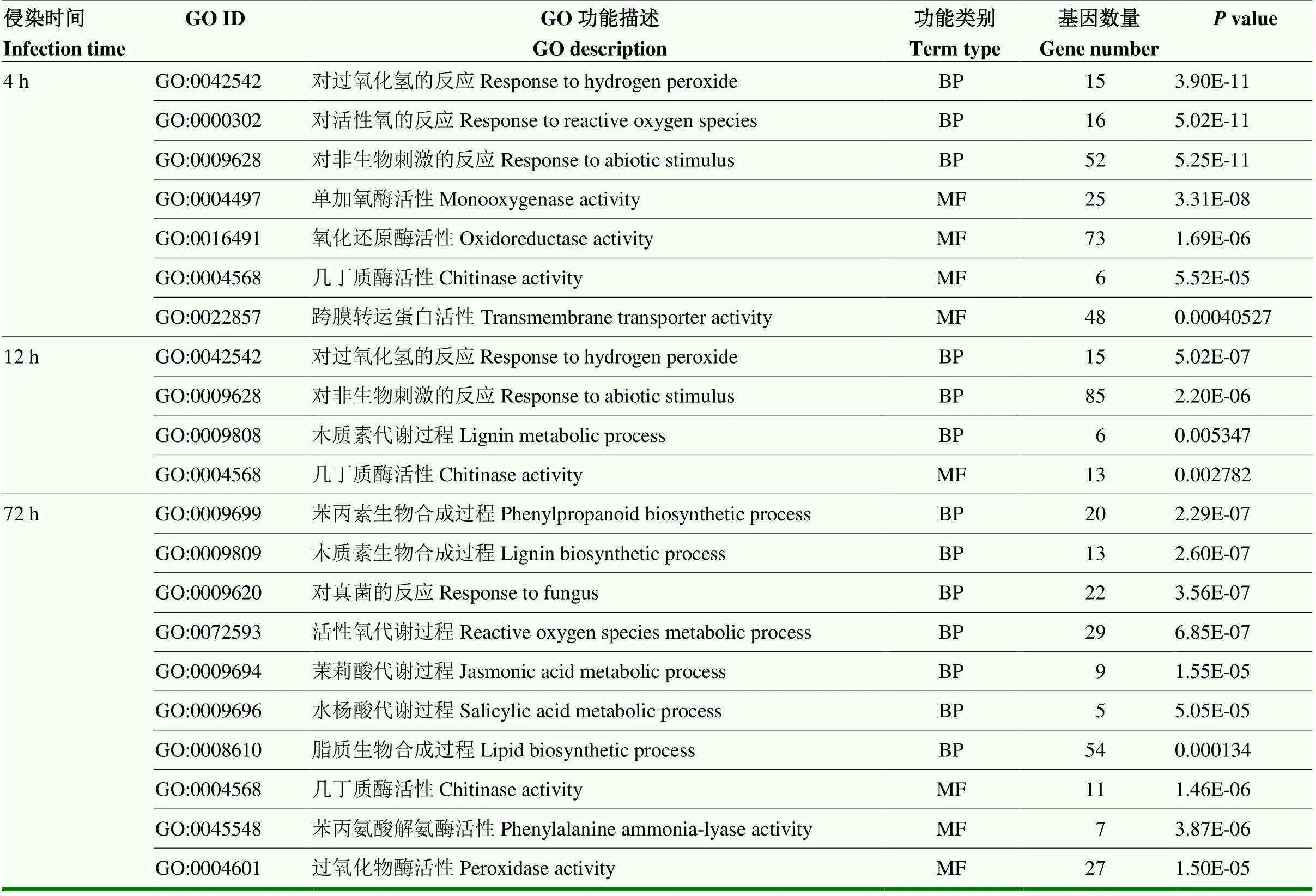
表4 玉米響應擬輪枝鐮孢侵染不同時間點差異表達基因的GO富集分類
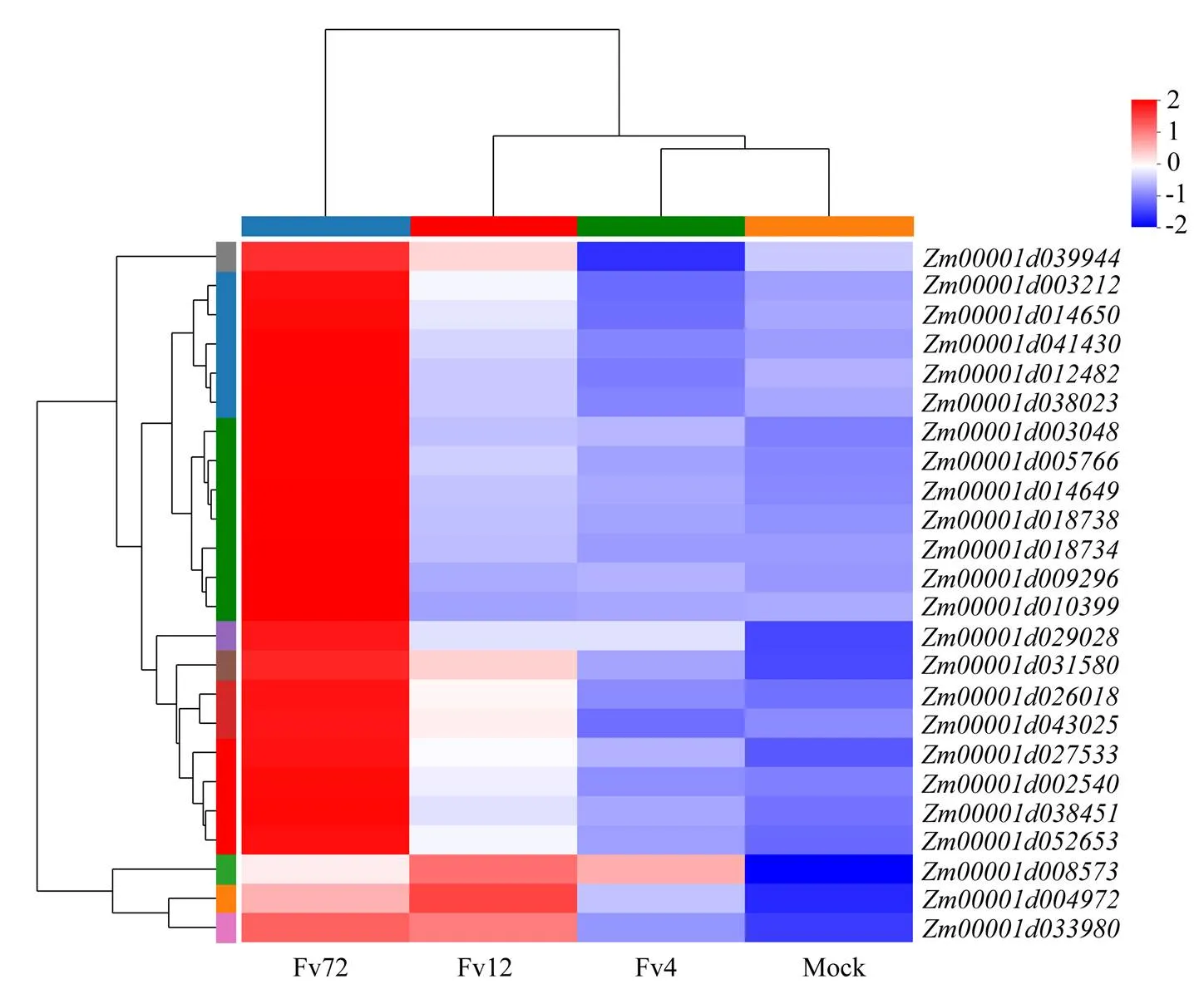
圖5 玉米中植物-抗病通路差異表達基因
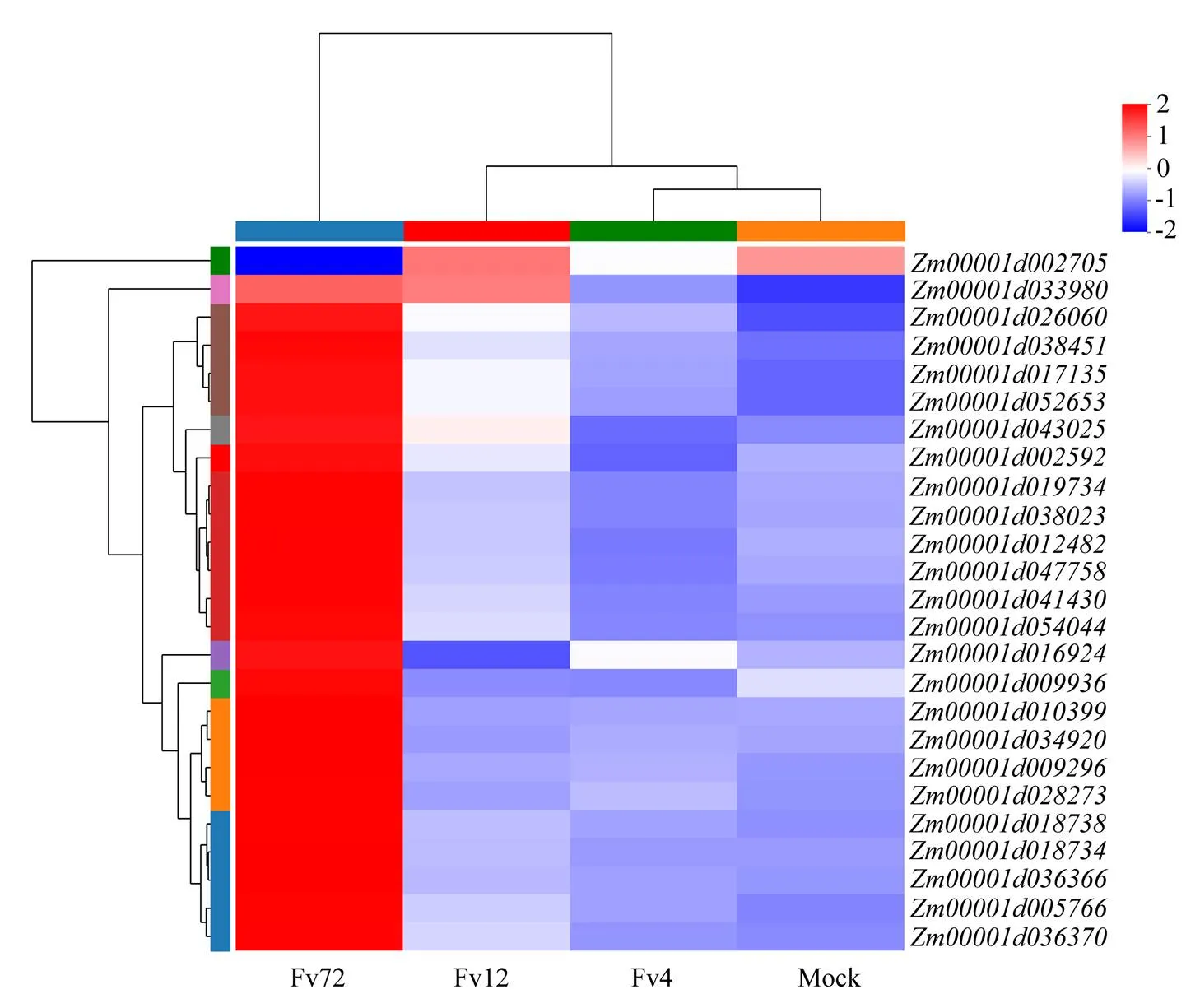
圖6 玉米中MAPK信號通路差異表達基因
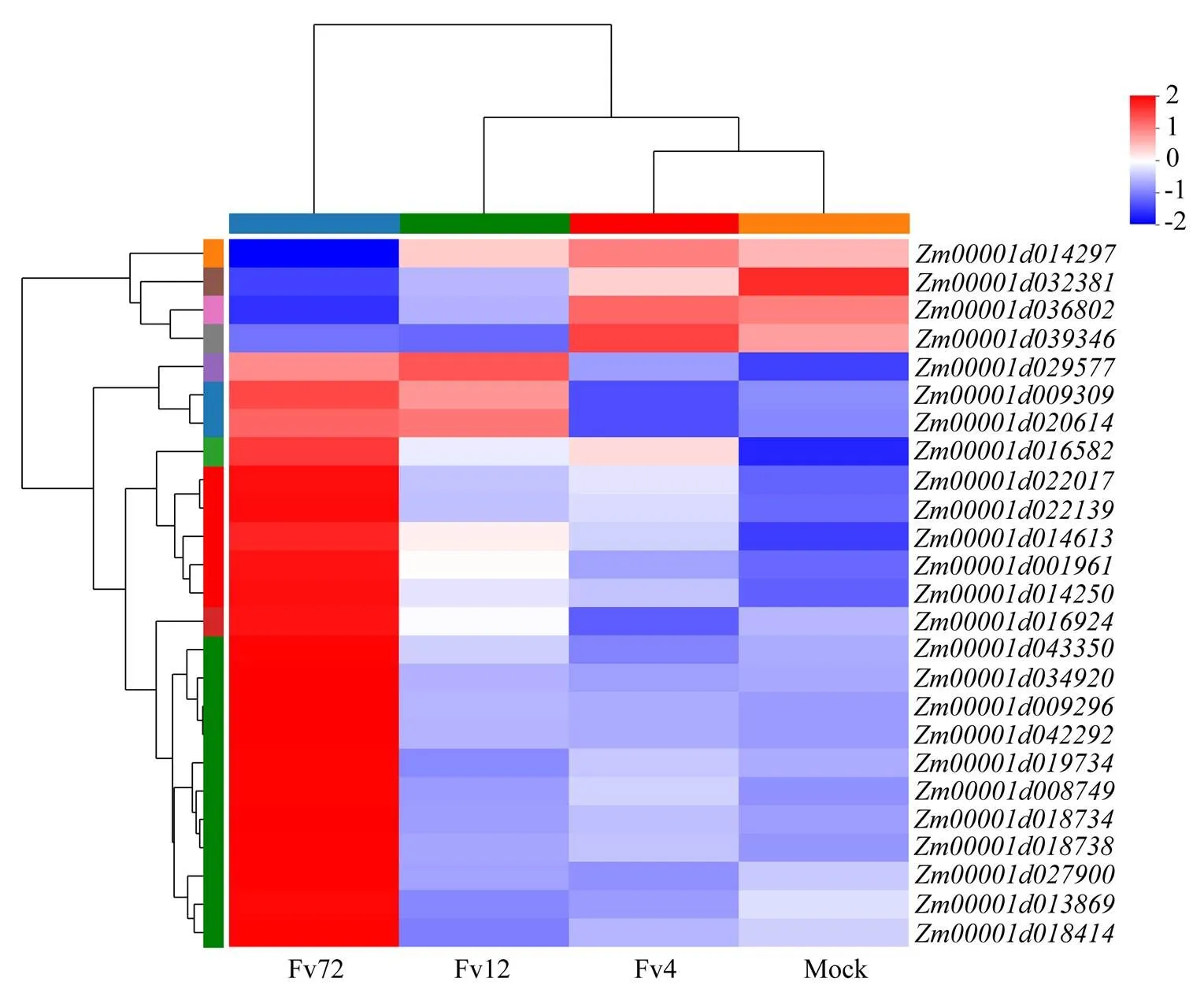
圖7 玉米中植物激素信號轉導途徑差異表達基因
2.5 差異基因的qRT-PCR驗證
利用qRT-PCR技術對隨機選取的6個玉米的差異表達基因和6個擬輪枝鐮孢的差異表達基因在接種擬輪枝鐮孢72 h后進行轉錄組數據驗證(圖8)。結果顯示,玉米響應病原菌侵染的植物-病原互作途徑中鈣依賴蛋白激酶CDPK基因(、),茉莉酸介導的信號通路中JAZ基因(),植物激素信號轉導途徑中GH3 基因(、)和MAPK信號途徑中WRKY 33轉錄因子()在接種擬輪枝鐮孢72 h后上調表達。在擬輪枝鐮孢中,參與脂質生物合成的,參與脂質轉運與代謝的羥基類固醇脫氫酶基因(),類固醇生物合成途徑中的和,幾丁質合酶基因()和淀粉與蔗糖代謝途徑中的在接種擬輪枝鐮孢72 h后上調表達。以上12個差異基因的表達趨勢和轉錄組測序結果一致,表明RNA-seq分析結果可靠。

圖8 qRT-PCR驗證擬輪枝鐮孢侵染玉米籽粒72 h后基因表達情況
3 討論
由擬輪枝鐮孢引起的玉米穗腐病嚴重影響玉米產量及品質,擬輪枝鐮孢產生的伏馬毒素對人、畜有毒害作用。由于化學農藥防治穗腐病效果不佳,玉米抗性種質資源相對匱乏,因此明確玉米-擬輪枝鐮孢互作中雙方的調控過程變得更加迫切。唐科志等[22]利用轉錄組高通量測序分析發現,紅橘被鏈格孢菌橘致病型(tangerine pathotype)侵染后,基礎代謝遭到嚴重破壞,病程相關蛋白(PR)上調表達響應病原菌侵染。乙烯防御信號通路差異基因顯著富集應對病原菌侵染。滕彩玲等[23]通過轉錄組測序分析,揭示了馬蜂柑響應黃龍病菌(‘Liberibacter asiaticus’)侵染不同時期的生物學過程。史毅等[24]利用轉錄組測序分析匍匐翦股穎對立枯絲核菌()的抗性,明確了病原菌侵染過程中寄主抑制病原菌擴散的抗病機制。
3.1 擬輪枝鐮孢侵染玉米籽粒的主要機制
在玉米-擬輪枝鐮孢互作過程中,對不同玉米抗/感品種響應病原菌侵染的轉錄分析較多[19-20],本文利用RNA高通量測序技術,對擬輪枝鐮孢侵染玉米不同時期進行了轉錄組測序,分析玉米和擬輪枝鐮孢雙方差異表達基因的功能。首先發現,侵染初期擬輪枝鐮孢只有少量差異基因主要富集在核糖體生物合成、細胞壁及脂肪酸生物合成途徑中。脂肪酸可以通過調控菌絲的生長和無性孢子的產生來調控真菌的生長和發育[25-27]。這表明與在培養基中生長的菌絲相比,剛進入植物中的擬輪枝鐮孢生長狀態與其相似,接菌后4 h仍在細胞間隙生長,還未侵入植物細胞內。但此時玉米的差異表達基因富集在活性氧、過氧化氫的反應等方面,表明擬輪枝鐮孢在細胞間隙的生長已經觸發了活性氧爆發。在植物免疫系統中,活性氧的產生是植物早期抗病反應之一,起著至關重要的作用,不僅直接降低微生物的生存能力,還產生觸發其他免疫反應的信號[28-30]。而且通過對植物-病原互作通路分析發現,CaMCML的基因、、在接菌后4 h上調表達,表明這種識別及反應是由細胞內Ca2+濃度發生快速變化引起的。
在接菌12 h后擬輪枝鐮孢開始出現大量的差異表達基因,富集在蛋白質和碳水化合物代謝、木聚糖酶活性、幾丁質結合相關通路,表明這段時間病菌生長旺盛,同時病菌產生降解植物細胞壁半纖維素結構的水解酶,從而破壞植物細胞。此時真菌侵染已經開始在玉米籽粒的細胞中定殖及生長[31],而作為觸發宿主PTI的細胞壁組分幾丁質合成也增加。此時玉米的差異表達基因進一步集中在苯丙素生物合成、木質素和幾丁質酶通路中,說明植物感知到真菌的侵染后通過木質化,形成病原菌入侵的第一道屏障,生物合成苯丙素等植物抗毒素。同時通過合成幾丁質酶降解真菌的細胞壁幾丁質成分,破壞擬輪枝鐮孢細胞壁的同時產生的低聚物誘發了玉米PTI,因此在后續72 h,玉米中包括植物-病原互作、MAPK途徑和植物激素信號轉導在內的多種途徑基因均表現為顯著上調。而接菌72 h后,擬輪枝鐮孢的差異基因主要富集在類固醇生物合成,淀粉和蔗糖代謝途徑中,碳水化合物和細胞壁多糖分解代謝過程,表明此時病菌開始大量利用植物細胞中的蔗糖和淀粉作為繁殖及進一步侵染的重要營養物質。此時,玉米中與苯丙素生物合成和代謝相關的基因上調。苯丙素可在植物中通過促進木質素沉積和類黃酮類植物抗毒素合成,對病原體感染作出反應[32-34]。
3.2 不同代謝通路介導的植物防御反應
在互作72 h時,玉米中包括植物-病原互作、MAPK途徑和植物激素信號轉導在內的抗病過程中大量差異基因較侵染前期相比顯著上調。MAPK信號級聯在調節植物生長發育和脅迫反應中發揮關鍵作用,包括植物防御激素的生物合成、信號傳遞、活性氧生成、氣孔關閉、防御基因激活、植保素生物合成、細胞壁強化和超敏反應(HR)細胞死亡[35]。與其他植物激素在抗病中的報道一樣,茉莉酸、水楊酸、乙烯等不同的激素信號通路也參與了植物與病原菌的互作,例如多個茉莉酸途徑關鍵基因JAZ、乙烯通路的EIN3和ERF1/2 基因。筆者發現在植物-病原互作途徑中,等多個及上調表達,表明它們參與了玉米的抗病。報道顯示,WRKY轉錄因子在植物抗病中發揮重要作用,而且可以調節PR1等編碼病程相關蛋白基因的表達以及茉莉酸、水楊酸等植物激素信號途徑[36-37]。擬南芥中,WRKY33基因缺失突變體對灰葡萄孢()和鏈格孢的敏感性增強,同時導致水楊酸水平升高而茉莉酸水平下調[38]。此外,BIRKENBIHL等[39]研究表明,WRKY33可直接靶向參與水楊酸、乙烯、茉莉酸等信號和鈣調素生物合成的基因。因此本研究中多個在接菌后持續上調可能在調節抗病相關過程中發揮了重要的作用。
4 結論
擬輪枝鐮孢先在玉米細胞間隙生長,后侵入細胞完成定殖,并利用宿主的營養物質進一步擴展。而玉米早期ROS爆發,后續通過幾丁質酶參與的PTI、木質化、抗毒素合成抵抗病菌侵入,最終MAPK、植物激素等多種抗病相關通路基因均被激活。互作雙方差異基因的挖掘為進一步研究抗擬輪枝鐮孢玉米穗腐病提供了依據。
[1] DUAN C X, QIN Z H, YANG Z H, LI W X, SUN S L, ZHU Z D, WANG X M. Identification of pathogenicspp. causing maize ear rot and potential mycotoxin production in China. Toxins, 2016, 8(6): 186.
[2] RHEEDER J P, MARASAS W F, VISMER H F. Production of fumonisin analogs byspecies. Applied and Environmental Microbiology, 2002, 68: 2101-2105.
[3] YAZDANPANAH H, SHEPHARD G S, MARASAS W F, VAN DER WESTHUIZEN L, RAHIMIAN H, SAFAVI S N, ESKANDARI P, GHIASIAN S A. Human dietary exposure to fumonisin B1 from Iranian maize harvested during 1998-2000. Mycopathologia, 2006, 161(6): 395-401.
[4] SUN G J, WANG S K, HU X, SU J J, HUANG T R, YU J H, TANG L L, GAO W M, WANG J S. Fumonisin B1 contamination of home- grown corn in high-risk areas for esophageal and liver cancer in China. Food Additives and Contaminants, 2007, 24(2): 181-185.
[5] GELDERBLOM W C A, SNYMAN S D. Mutagenicity of potentially carcinogenic mycotoxins produced by. Mycotoxin Research, 1991, 7: 46-52.
[6] WILSON R A.. Trends in Microbiology, 2021, 29(7): 663-664.
[7] PRITSCH C, MUEHLBAUER G J, BUSHNELL W R, SOMERS D A, VANCE C P. Fungal development and induction of defense response genes during early infection of wheat spikes by. Molecular Plant-Microbe Interactions, 2000, 13(2): 159-169.
[8] AN H J, LURIE S, GREVE L C, ROSENQUIST D, KIRMIZ C, LABAVITCH J M, LEBRILLA C B. Determination of pathogen- related enzyme action by mass spectrometry analysis of pectin breakdown products of plant cell walls. Analytical Biochemistry, 2005, 338: 71-82.
[9] PEKKARINEN A, MANNONEN L, JONES B L, NIKU-PAAVOLA M L. Production of proteases byspecies grown on barley grains and in media containing cereal proteins. Journal of Cereal Science, 2000, 31(3): 253-261.
[10] WANYOIKE W M, KANG Z, BUCHENAUER H. Importance of cell wall degrading enzymes produced byduring infection of wheat head.European Journal of Plant Pathology, 2002, 108: 803-810.
[11] JONES J D, DANGL J L. The plant immune system. Nature, 2006, 444(7117): 323-329.
[12] MILLER G, SHULAEV V, MITTLER R. Reactive oxygen signaling and abiotic stress. Physiologia Plantarum, 2008, 133(3): 481-489.
[13] GADJEV I, VANDERAUWERA S, GECHEV T S, LALOI C, MINKOV I N, SHULAEV V, Apel K, INZE D, MITTLER R, BREUSEGEM F V. Transcriptomic footprints disclose specificity of reactive oxygen species signaling in. Plant Physiology, 2006, 141(2): 436-445.
[14] BARI R, JONES J D. Role of plant hormones in plant defence responses. Plant Molecular Biology, 2009, 69(4): 473-488.
[15] NAVARRO L, BARI R, ACHARD P, LISON P, NEMRI A, HARBERD N P, JONES J D G. Dellas control plant immune responses by modulating the balance of jasmonic acid and salicylic acid signaling. Current Biology, 2008, 18(9): 650-655.
[16] AMIL-RUIZ F, GARRIDO-GALA J, GADEA J, BLANCO- PORTALES R, MUNOZ-MERIDA A, TRELLES O, DE LOS SANTOS B, ARROYO F T, AGUADO-PUIG A, ROMERO F, MERCADO J A, PLIEGO-ALFARO F, MUNOZ-BLANCO J, CABALLERO J L. Partial activation of SA- and JA-defensive pathways in strawberry uponinteraction. Frontiers in Plant Science, 2016, 7: 1036.
[17] GORMAN Z, CHRISTENSEN S A, YAN Y, HE Y, BORREGO E, KOLOMIETS M V. Green leaf volatiles and jasmonic acid enhance susceptibility to anthracnose diseases caused byin maize. Molecular Plant Pathology, 2020, 21(5): 702-715.
[18] 劉俊. 擬輪枝鐮孢和層出鐮孢侵染玉米果穗的途徑及鐮孢菌病害防治[D]. 保定: 河北農業大學, 2017.
LIU J. The pathway of infecting ear byandanddisease control[D]. Baoding: Hebei Agricultural University, 2017. (in Chinese)
[19] WANG Y P, ZHOU Z J, GAO J Y, WU Y B, XIA Z L, ZHANG H Y, WU J Y. The mechanisms of maize resistance toby comprehensive analysis of RNA-seq data. Frontiers in Plant Science, 2016, 7: 1654.
[20] LANUBILE A, FERRARINI A, MASCHIETTO V, DELLEDONNE M, MAROCCO A, BELLIN D. Functional genomic analysis of constitutive and inducible defense responses toinfection in maize genotypes with contrasting ear rot resistance. BMC Genomics, 2014, 15(1): 710.
[21] SHU X M, LIVINGSTON D P, FRANKS R G, BOSTON R S, WOLOSHUK C P, PAYNE G A. Tissue-specific gene expression in maize seeds during colonization byand. Molecular Plant Pathology, 2015, 16(7): 662-674.
[22] 唐科志, 周常勇. 紅橘響應褐斑病菌侵染的轉錄組學分析. 中國農業科學, 2020, 53(22): 4584-4600.
TANG K Z, ZHOU C Y. Transcriptome analysis ofBlanco, cv. Hongjv infected withtangerine pathotype. Scientia Agricultura Sinica, 2020, 53(22): 4584-4600. (in Chinese)
[23] 滕彩玲, 鐘晰, 吳昊娣, 胡燕, 周常勇, 王雪峰. 馬蜂柑響應黃龍病菌不同侵染時期的生物學和轉錄組學分析. 中國農業科學, 2020, 53(7): 1368-1380.
TENG C L, ZHONG X, WU H D, HU Y, ZHOU C Y, WANG X F. Biologic and transcriptomic analysis ofresponses to ‘Liberibacter asiaticus’ at different infection stages. Scientia Agricultura Sinica, 2020, 53(7): 1368-1380. (in Chinese)
[24] 史毅, 牛奎舉,馬暉玲. 匍匐翦股穎接種立枯絲核菌后基因表達變化的轉錄組學分析. 中國農業科學, 2017, 50(17): 3323-3336.
SHI Y, NIU K J, MA H L. Transcriptome analysis of creeping bentgrass () infected with. Scientia Agricultura Sinica, 2017, 50(17): 3323-3336. (in Chinese)
[25] CALVO A M, HINZE L L, GARDNER H W, KELLER N P. Sporogenic effect of polyunsaturated fatty acids on development ofspp.. Applied and Environmental Microbiology, 1999, 65(8): 3668-3673.
[26] TSITSIGIANNIS D I, KOWIESKI T M, ZARNOWSKI R, KELLER N P. Three putative oxylipin biosynthetic genes integrate sexual and asexual development in. Microbiology, 2005, 151(6): 1809-1821.
[27] 晏石娟, 黃文潔, 劉春明. 脂肪酸及其氧合物對曲霉屬真菌菌絲生長、產孢和黃曲霉毒素合成的影響. 微生物學報, 2017, 57(1): 24-32.
YAN S J, HUANG W J, LIU C M. Effects of fatty acids and oxylipins on fungal growth, sporulation and aflatoxin production in. Acta Microbiologica Sinica, 2017, 57(1): 24-32. (in Chinese)
[28] BIGEARD J, COLCOMBET J, HIRT H. Signaling mechanisms in pattern-triggered immunity (PTI). Molecular Plant, 2015, 8(4): 521-539.
[29] MITTLER R. ROS are good.2017, 22: 11-19.
[30] TSUDA K, KATAGIRI F. Comparing signaling mechanisms engaged in pattern-triggered and effector-triggered immunity. Current Opinion in Plant Biology, 2010, 13(4): 459-465.
[31] JIA L J, TANG H Y, WANG W Q, YUAN T L, WEI W Q, PANG B, GONG X M, WANG S F, LI Y J, ZHANG D, LIU W, TANG W H. A linear nonribosomal octapeptide fromfacilitates cell-to-cell invasion of wheat. Nature Communications, 2019, 10(1): 922.
[32] DONG N Q, LIN H X. Contribution of phenylpropanoid metabolism to plant development and plant-environment interactions. Journal of Integrative Plant Biology, 2021, 63(1): 180-209.
[33] LANUBILE A, MASCHIETTO V, BORRELLI V M, STAGNATI L, LOGRIECO A F, MAROCCO A. Molecular basis of resistance toear rot in maize.,2017, 8: 1774.
[34] ZHANG Y, WU L, WANG X, CHEN B, ZHAO J, CUI J, LI Z, YANG J, WU L, WU J, ZHANG G, MA Z. The cotton laccase geneenhanceswilt resistance via an increase in defence- induced lignification and lignin components in the cell walls of plants., 2019, 20(3): 309-322.
[35] MENG X, ZHANG S. MAPK cascades in plant disease resistance signaling. Annual Review of Phytopathology, 2013, 51: 245-266.
[36] LIAO C J, LAI Z, LEE S, YUN D J, MENGISTE T.HOOKLESS1 regulates responses to pathogens and abscisic acid through interaction with MED18 and acetylation of WRKY33 and ABI5 chromatin. ThePlant Cell, 2016, 28(7): 1662-1681.
[37] ZHANG X, MENARD R, LI Y, CORUZZI G M, HEITZ T, SHEN W H, BERR A.SDG8 potentiates the sustainable transcriptional induction of the pathogenesis-related genesandduring plant defense response. Frontiers in Plant Science, 2020, 11: 277.
[38] ZHENG Z, QAMAR S A, CHEN Z, MENGISTE T.WRKY33 transcription factor is required for resistance to necrotrophic fungal pathogens. The Plant Journal, 2006, 48(4): 592-605.
[39] BIRKENBIHL R P, DIEZEL C, SOMSSICH I E.WRKY33 is a key transcriptional regulator of hormonal and metabolic responses towardinfection. Plant Physiology, 2012, 159(1): 266-285.
Screening of Differential Genes and Analysis of Metabolic Pathways in the Interaction betweenand Maize Kernels
QU Qing1,2, LIU Ning2,3, ZOU JinPeng2,3, ZHANG YaXuan3, JIA Hui3, SUN ManLi2,3, CAO ZhiYan2,3, DONG JinGao2,3
1College of Life Sciences, Hebei Agricultural University, Baoding 071001, Hebei;2Hebei Key Laboratory ofPlant Physiology and Molecular Pathology/State Key Laboratory of North China Crop Improvement and Regulation, Baoding 071001, Hebei;3College of Plant Protection, Hebei Agricultural University, Baoding 071001, Hebei
【Objective】 Maize ear rot caused byis one of the most serious diseases in maize producing areas in China. The objective of this study is to understand the differences in gene expression during the plant-pathogen interaction at different stages, and to provide a basis for pathogenic mechanism of the pathogen infection and resistance mechanism of maize.【Method】Illumina platform was used to sequence the transcriptome of maize kernels infected withat0, 4, 12, and 72 h.The differentially expressed genes (DEGs) of maize andwere screened with |log2FC|≥1,-adjust<0.05 as threshold and clean reads were compared with genome of maize and, separately. Functional annotation and enrichment analysis of DEGs were carried out by using GO and KEGG databases. Goatools software was used to analyze the expression changes of genes related to plant-pathogen interaction, MAPK signaling pathway and plant hormone signal transduction pathway. Sequencing results were verified by quantitative real-time PCR (qRT-PCR).【Result】A total of 140, 400 and 1 945 DEGs were up-regulated and 9, 302, and 1 784 DEGs were down-regulated inafter 4, 12 and 72 h interaction, respectively. A total of 293, 692, and 1 426 DEGs were up-regulated and 320, 482, and 153 DEGs were down-regulated in maize after 4, 12 and 72 h interaction, respectively. GO and KEGG enrichment analysis of DEGs showed thatgrew in intercellular space at the early stage of pathogen infection. The DEGs were enriched in RNA biosynthesis, cell wall structural component, fatty acid biosynthesis, protein metabolism, carbohydrate metabolism, biological process, and metabolic process. reactive oxygen species (ROS) was triggered in maize at the early stage of infection. The DEGs were enriched in response to ROS, hydrogen peroxide, chitinase activity, monooxygenase activity, lignin metabolism. At the later stage of infection,colonized and expanded in maize, and the DEGs were enriched in carbohydrate and cell wall polysaccharide catabolic process, transmembrane transport and oxidoreductase activity. Maize responded to pathogen infection through phenylpropanoid, lignin, flavonoid biosynthesis, MAPK signaling pathway, plant-pathogen interaction and plant hormone signal transduction. Six DEGs of maize and six DEGs ofwere randomly selected for qRT-PCR. The results were consistent with those of transcriptome sequencing, which confirmed the accuracy of RNA-seq.【Conclusion】at the early stage of infection,grew in the intercellular space, triggering ROS outbreak in maize and the expression of related pathway differential genes. At the middle and late stages of infection, the pathogen further colonized and expanded in maize with starch as nutrient. Maize responded to the infection ofthrough biosynthesis of phenylpropanoid, lignin and chitinase. Meanwhile, plant-pathogen interaction, MAPK signaling pathway, and plant hormone signal transduction were involved in the resistance to the infection of.
; maize ear rot; transcriptome; plant-pathogen interaction; gene expression; differentially expressed gene (DEG)

10.3864/j.issn.0578-1752.2023.06.006
2022-10-27;
2022-11-23
國家玉米產業技術體系(CARS-02)、華北作物改良與調控國家重點實驗室自主科研項目(NCCIR2020ZZ-12)、河北省重點研發計劃(20326502D)
渠清,E-mail:qu_qing@126.com。通信作者曹志艷,E-mail:caozhiyan@hebau.edu.cn。通信作者董金皋,E-mail:shmdjg@hebau.edu.cn
(責任編輯 岳梅)
猜你喜歡
少兒科學周刊·兒童版(2021年19期)2021-12-10 14:13:40
小學閱讀指南·低年級版(2021年3期)2021-03-19 06:12:40
小天使·二年級語數英綜合(2020年8期)2020-12-23 04:57:40
小天使·一年級語數英綜合(2020年11期)2020-12-16 02:57:22
學苑創造·A版(2020年3期)2020-04-24 09:21:39
小溪流(畫刊)(2017年11期)2018-01-09 19:15:14
少兒科學周刊·兒童版(2017年5期)2017-06-29 22:24:28
少兒科學周刊·兒童版(2017年5期)2017-06-29 16:46:33
紅領巾·萌芽(2017年5期)2017-06-23 10:35:59
爆笑show(2016年7期)2017-02-09 09:36:13
