肝纖維化治療的新視角:靶向巨噬細胞代謝
2023-04-29 21:39:05許鈞金衛林李汛
臨床肝膽病雜志 2023年4期
許鈞 金衛林 李汛
摘要:肝纖維化是由多種病因引起的細胞外基質過度累積,導致纖維瘢痕形成的病理過程。目前,病因治療(如有效的抗病毒治療)仍是肝纖維化的主要治療策略。肝巨噬細胞作為肝纖維化的核心參與者,影響肝纖維化的進展與消退,被認為是肝纖維化治療的重要靶點。近年來,隨著免疫代謝領域的進展,代謝重編程已成為決定巨噬細胞結局和推動疾病發展的關鍵因素。本文綜述了肝纖維化過程中巨噬細胞的作用及其代謝變化,并討論靶向巨噬細胞代謝的抗纖維化潛力,為肝纖維化的發生、發展和治療提供新的思路
關鍵詞:肝硬化; 巨噬細胞; 治療學
基金項目:國家自然科學基金地區項目(82060119)
A new perspective in the treatment of liver fibrosis: Targeting macrophage metabolism
XU Jun1, JIN Weilin1,2b, LI Xun1,2a,2b,3. (1. The First Clinical Medical College of Lanzhou University, Lanzhou 730000, China; 2. a. Medical Frontier Innovation Research Center, b. Department of General Surgery, The First Hospital of Lanzhou University, Lanzhou 730000, China; 3. Gansu Province Key Laboratory of Biotherapy and Regenerative Medicine, Lanzhou 730000, China)
Corresponding author:
LI Xun, Lxdr21@126.com (ORCID:0000-0003-3787-1558)
Abstract:
Liver fibrosis is a pathological process of fibrous scar formation caused by excessive accumulation of extracellular matrix due to various etiologies. At present, etiological treatment, such as effective antiviral therapy, is still the main treatment strategy for liver fibrosis. As the core participant of liver fibrosis, liver macrophages affect the progression and regression of liver fibrosis and are thus considered an important target for the treatment of liver fibrosis. With the recent advances in the field of immune metabolism, metabolic reprogramming has become a key factor for determining the outcome of macrophages and promoting the development of diseases. This article reviews the role and metabolic changes of macrophages during liver fibrosis and discusses the anti-fibrotic potential of targeting macrophage metabolism, so as to provide new ideas for the development, progression, and treatment of liver fibrosis.
Key words:Liver Cirrhosis; Macrophages; Therapeutics
Research funding:
Regional Project of National Natural Science Foundation of China (82060119)
肝纖維化是由病毒性肝炎、膽汁淤積性肝病、酒精性肝病、非酒精性脂肪性肝病、藥物及毒素等多種病因引起的持續性肝實質慢性炎癥,最終導致肝細胞外基質過度沉積,形成纖維性瘢痕。若不及時干預,肝纖維化最終發展為肝硬化甚至肝細胞癌,并出現一系列嚴重并發癥,如肝衰竭、門靜脈高壓癥、肝性腦病等[1]。肝臟富含巨噬細胞,包括常駐巨噬細胞和單核細胞來源的巨噬細胞(monocyte-derived macrophage, MDM)。巨噬細胞作為肝臟中重要的免疫細胞,其激活、極化和生物功能受到自身新陳代謝的調控[2]。近年來,隨著對免疫細胞代謝的深入研究發現,免疫細胞的代謝途徑及代謝產物在其激活、分化中發揮重要調節作用[3]。本文將對肝纖維化過程中巨噬細胞的作用及其代謝變化進行綜述,并討論靶向巨噬細胞代謝的抗纖維化潛力,為肝纖維化治療提供新的思路。
1 肝臟巨噬細胞的起源及分型
肝臟巨噬細胞占人體總巨噬細胞的90%,具有顯著異質性,主要由肝臟駐留巨噬細胞和各種浸潤巨噬細胞組成[4]。肝臟駐留巨噬細胞,又稱Kupffer細胞,通常存在于肝竇中,其起源于卵黃囊衍生的特定祖細胞,在胚胎發生期間已定植在肝臟組織中,也可通過骨髓來源的單核細胞分化進行補充[5]。Kupffer細胞在肝臟中自我更新,可清除病原體,吞噬細胞碎片和調節鐵代謝等,維持肝臟穩態。浸潤巨噬細胞主要包括骨髓來源的巨噬細胞、腹膜巨噬細胞和脾巨噬細胞[6]。其中,骨髓來源的巨噬細胞是浸潤巨噬細胞的主要成員,主要在肝臟病理狀態下發揮功能,在Kupffer細胞和肝星狀細胞(hepatic stellate cell, HSC)活化后募集,是肝巨噬細胞耗竭后補充和再生的重要來源。當發生肝損傷時,除骨髓來源的巨噬細胞外,腹膜巨噬細胞完成自我更新,積聚在包膜下的肝臟組織中,通過間皮細胞遷移促進肝臟再生。此外,由于脾臟是單核細胞儲存和分布的部位,脾巨噬細胞在肝損傷時也可被招募到肝臟發揮免疫調節作用[7]。總的來說,在正常肝臟中,Kupffer細胞作為肝臟的前哨細胞,占據肝巨噬細胞主導地位,維持肝臟穩態。當肝臟受外部因素影響形成病變時,Kupffer細胞首先接收信號并分化成不同的表型,產生促炎或抗炎因子,同時募集大量其他巨噬細胞到肝臟,即MDM。MDM具有與Kupffer細胞相似的功能和可塑性,在肝臟疾病的進展和逆轉中起重要作用[8]。
巨噬細胞根據激活途徑、細胞表面標志物及其釋放的細胞因子可分為兩種類型:經典活化(M1)型巨噬細胞和替代活化(M2)型巨噬細胞[9]。M1型巨噬細胞又稱為促炎型巨噬細胞,主要由脂多糖和IFNγ誘導激活,參與Th1及Th17型免疫反應。M1型巨噬細胞可分泌IL-1β、IL-6、IL-12、IL-23、TNFα等細胞因子,發揮抗原遞呈功能,還具有促進炎癥、清除病原微生物和抗腫瘤的生物學功能。M1型巨噬細胞極化的常見機制包括:(1)TLR4/NF-κB信號通路。目前多種藥物(如小檗堿、槲皮素)已被報道可以通過抑制TLR4/NF-κB信號通路來抑制M1型巨噬細胞極化[10-11]。(2)JAK/STAT1信號通路。IFNγ與其受體結合后激活JAK,誘導STAT1的磷酸化,從而使M1型巨噬細胞極化[12]。(3)Notch信號通路。研究[13]表明M1型巨噬細胞Notch受體表達顯著增加,可以通過靶向Notch信號通路來調節M1型巨噬細胞極化[14-15]。M2型巨噬細胞則被稱為抑炎型巨噬細胞,主要由IL-4和IL-13誘導激活,參與Th2型免疫反應。M2型巨噬細胞可分泌抗炎因子,如IL-10、IL-4、IL-13、TGFβ等,具有抑制炎癥,促進組織重塑,預防寄生蟲感染以及參與血管生成、免疫調節等生物學功能[16]。M2型巨噬細胞極化的常見機制包括:(1)JAK/STAT6信號通路。JAK/STAT6是M2型巨噬細胞極化的重要途徑,姜黃素通過分泌IL-4和IL-13上調STAT6表達,從而誘導M2型巨噬細胞極化[17]。(2)TGFβ/Smads信號通路。槲皮素通過抑制TGFβ1-Smad2/3途徑來抑制M2型巨噬細胞極化[18]。
巨噬細胞不同極化表型在肝臟疾病的發生和進展中發揮不同作用,對肝臟疾病具有雙重調節作用[19]。HSC是肝纖維化的主要效應細胞,靜止的HSC轉化為肌成纖維細胞是肝纖維化發病機制的核心,而巨噬細胞在HSC活化的過程中起關鍵作用[1]。由此,肝巨噬細胞在肝纖維化中發揮雙重作用,既可促進纖維化進展,也可使纖維化消退[20]。
2 肝臟巨噬細胞在肝纖維化中的雙重作用
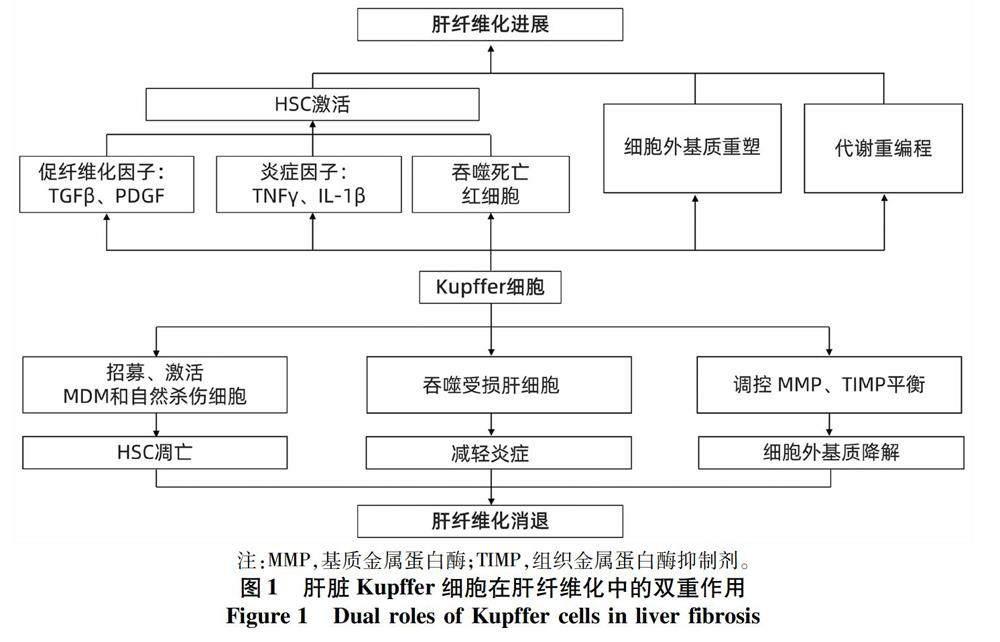
2.1 M1型巨噬細胞推動肝纖維化進展 在肝纖維化的起始和進展階段,損傷相關的模式分子及凋亡小體激活Disse空間中的Kupffer細胞并使其表型發生轉變,此時肝臟巨噬細胞表型以M1型(促炎型)為主。激活的Kupffer細胞可通過如圖1所示的方式促進肝纖維化的發生及發展:(1)Kupffer細胞可產生TGFβ、血小板生長因子(platelet-derived growth factor, PDGF)等促纖維化因子,激活HSC,使其向肌成纖維細胞轉化,促進肝纖維化發生[21];(2)Kupffer細胞產生TNFγ、IL-1β等炎癥因子,招募肝外炎癥細胞入肝,進一步加重肝細胞損傷。同時,炎癥因子可通過NF-κB途徑激活HSC,并維持HSC活性,促進肝纖維化進展[22];(3)Kupffer細胞表達MMP,如MMP-9,促進細胞外基質沉積[23];(4)激活的Kupffer細胞吞噬死亡的紅細胞,使來源于血紅蛋白中的鐵沉積在肝臟,誘導氧化應激與炎癥反應,進而激活HSC,促進纖維化發生[24];(5)活化的Kupffer細胞可破壞肝臟血管結構,使肝臟局部環境缺氧,從而觸發缺氧誘導的肝纖維化形成[25]。
除Kupffer細胞,肝臟中MDM浸潤也影響肝纖維化的發生。小鼠肝臟損傷期間Ly6C高表達(Ly6Chigh)單核巨噬細胞可促進炎癥的發生;相反,Ly6C低表達(Ly6Clow)單核巨噬細胞具有抗炎作用[26]。在肝臟損傷早期,Ly6Chigh單核巨噬細胞通過趨化因子配體2(chemokine ligand-2, CCL2)/趨化因子受體2(C-C chemokine receptor, CCR2)軸的作用,被募集到損傷肝臟中,釋放TGFβ、IL-1b、PDGF、CCL2等作用于HSC,使其活化、增殖,從而推動肝纖維化發生[27]。在人體中,MDM主要包括CD14++CD16-單核巨噬細胞、CD14++CD16+單核巨噬細胞和CD14+CD16++單核巨噬細胞3種類型,其中CD14++CD16+單核巨噬細胞是參與肝纖維化形成的主要細胞類型。當肝損傷時,CD14++CD16+單核細胞在受損的肝臟內聚集,釋放炎癥因子和纖維化因子,促進肝纖維化形成[28]。
2.2 M2型巨噬細胞促進肝纖維化消退 在肝纖維化消退過程中,肝臟巨噬細胞表型以M2型(抑炎型)為主,Kupffer細胞可能通過以下方式發揮作用:(1)Kupffer細胞招募并激活MDM和自然殺傷細胞,使活化HSC凋亡,從而發揮抗纖維化作用[29];(2)Kupffer細胞可吞噬損傷的肝細胞,減輕炎癥反應,從而減緩肝纖維化的進程。(3)Kupffer細胞在提高MMP(如 MMP-1、MMP-13)表達的同時,可調節MMP與TIMP的平衡,降解細胞外基質,促進肝纖維化消退[29-30]。
在小鼠肝臟內Ly6Clow單核巨噬細胞通過下調TGFβ等促炎因子水平,減輕肝臟炎癥,誘導活化的HSC衰老、死亡。同時Ly6Clow單核巨噬細胞中部分MMP(如MMP-12、MMP-13)表達水平升高,參與細胞外基質降解[27]。
3 肝臟巨噬細胞代謝重編程與肝纖維化
肝損傷后,肝臟的局部微環境發生變化,為應對微環境的變化,巨噬細胞的代謝方式及代謝產物發生變化,即代謝重編程。肝臟巨噬細胞的代謝重編程是影響巨噬細胞極化、HSC活化以及肝纖維化發展和消退的重要因素[31]。M1型巨噬細胞的代謝特征包括糖酵解、磷酸戊糖途徑的增強和三羧酸循環的激活(生成用于脂肪酸合成的檸檬酸)。M2型巨噬細胞的代謝特征包括脂肪酸氧化、精氨酸酶途徑的增強以及三羧酸循環的激活(參與氧化磷酸化)[3]。在此部分中,筆者將討論細胞內營養物質代謝的變化如何影響肝纖維化中的巨噬細胞極化和功能。
3.1 巨噬細胞葡萄糖代謝與肝纖維化 肝臟急、慢性損傷及繼發炎癥時,多伴隨乏氧現象,乏氧微環境作為肝臟物理化學損傷的重要因素之一,對巨噬細胞的激活和肝纖維化的形成與發展有重要作用[25]。在缺氧條件下,巨噬細胞經歷從氧化磷酸化到有氧糖酵解的代謝轉換,以滿足其高能量需求。該過程受缺氧誘導因子1α調控,糖酵解關鍵蛋白(葡萄糖轉運體1)以及糖酵解的關鍵酶(如己糖激酶、丙酮酸激酶和果糖-2,6-二磷酸酶3)的表達增加[32]。此時巨噬細胞表型以M1型為主,推動纖維化的發生及發展。除了增強的糖酵解外,M1型巨噬細胞中不完整的三羧酸循環,導致琥珀酸鹽和檸檬酸鹽積累;過量的琥珀酸鹽使缺氧誘導因子1α更加穩定,這反過來又維持了M1型巨噬細胞的糖酵解代謝,從而增強M1型巨噬細胞中炎性因子IL-1β的分泌[33]。
3.2 巨噬細胞脂質代謝與肝纖維化 脂肪酸合成增強促使巨噬細胞極化為M1型。在巨噬細胞中飽和脂肪酸的結合或氧化脂蛋白被清除受體(如巨噬細胞清除受體1)捕獲,導致M1型巨噬細胞的形成[34]。與上述結論一致,缺乏巨噬細胞清除受體1的小鼠在高脂肪飲食中表現出更少的肝臟炎癥和更強的抗纖維化能力[35]。此外,有研究[36]表明,巨噬細胞暴露于脂肪酸中可導致具有細胞毒性的脂質(如二酰基甘油和神經酰胺)積累,使巨噬細胞進入促炎狀態。
3.3 巨噬細胞微量元素代謝與肝纖維化? 鐵代謝失調和肝鐵超載與非酒精性脂肪性肝炎(non-alcoholic steatohepatitis, NASH)及晚期肝纖維化有關。在高脂飲食誘導的NASH模型中,具有促炎表型的富鐵Kupffer細胞通過激活MiT/TFE轉錄因子促進NASH相關肝纖維化的發展[37]。鐵超載可破壞巨噬細胞M1/M2極化的平衡,誘導表型轉變為M1型,導致低脂飲食小鼠的炎癥和纖維生成[38]。
4 靶向巨噬細胞免疫代謝的抗纖維化治療
鑒于巨噬細胞在肝纖維化發生發展中的重要作用以及免疫細胞代謝重編程機制的闡明,靶向免疫代謝重編程可能是肝纖維化的一種潛在治療策略,以下將介紹肝纖維化中巨噬細胞的一些代謝靶點。
轉錄因子C-Rel是協調巨噬細胞糖代謝,誘導巨噬細胞極化的關鍵角色。當轉錄因子C-Rel與表達6-磷酸果糖-2-激酶-3的啟動子結合時,細胞糖酵解增強,誘導巨噬細胞M1型極化,最終導致肝損傷模型中的HSC激活,推動肝纖維化發生;當轉錄因子C-Rel與6-磷酸果糖-2-激酶-1的啟動子結合時,細胞的氧化磷酸化增強,誘導巨噬細胞的M2型極化,抑制HSC活化,從而抑制肝纖維化的發生及發展[39]。此外,膜聯蛋白家族成員膜聯蛋白A5,通過與丙酮酸激酶 M2相互作用,使肝巨噬細胞從糖酵解轉變為氧化磷酸化,這種代謝重編程過程刺激肝巨噬細胞的激活和從M1型到M2型的表型轉換,從而改善高脂飲食NASH 模型中的脂肪變性、炎癥和肝纖維化[40]。
巨噬細胞中脂肪酸合成增強導致巨噬細胞極化為M1型,推動肝纖維化的發生。脂肪酸合成的限速步驟是乙酰輔酶A轉化為丙二酰輔酶 A,此過程由乙酰輔酶A羧化酶 (acetyl-CoA carboxylase, ACC)催化。Gao等[41]通過使用ACC抑制劑WZ66,可抑制ACC進而減輕肝脂肪變性,阻止巨噬細胞的活化和浸潤,降低HSC的活化[42]。同時在一項ACC抑制劑對NASH患者的Ⅱ期臨床試驗[43]中,也有類似結果報道。
過氧化物酶體增殖物激活受體(peroxisome proliferator-activated receptor, PPAR)是一個核轉錄因子家族,包括α、β、δ和γ四種亞型,參與脂質代謝和葡萄糖代謝的調節。PPARα亞型主要存在于肝細胞中,少量存在于巨噬細胞和內皮細胞。PPARβ/δ亞型在所有的肝臟細胞中表達,而PPARγ亞型在巨噬細胞和靜止的HSC中表達[44]。已有研究[45]發現PPARα的定位在肝臟炎癥期間會從肝細胞轉移到Kupffer細胞上,而PPARα的激活可使巨噬細胞表型轉變為M2型。目前在野生小鼠以及蛋氨酸和膽堿缺乏飲食飼養的小鼠中發現PPARα激動劑(WY-14643)可以激活PPARα,減輕肝臟脂質積累和肝臟炎癥,從而減緩肝纖維化發生[46-47]。PPARβ/δ盡管在Kupffer細胞中表現出抗炎特性,但對HSC具有活化作用,其激動劑(GW501516)的肝保護和抗纖維化作用有待在臨床試驗中得到證實[48]。PPARγ亞型可下調炎癥基因的表達,使巨噬細胞轉變為M2型[48]。雖然在使用PPARγ激動劑(如吡格列酮或羅格列酮)的動物模型中纖維化改善明顯[49],但在羅格列酮治療1~2年后,患者肝纖維化并未得到改善[50]。
法尼醇X受體(farnesoid X receptor, FXR)是一種膽汁酸受體,主要存在于肝細胞、Kupffer細胞、肝竇內皮細胞和HSC中。FXR通過誘導脂蛋白代謝相關基因表達,抑制與肝臟甘油三酯合成相關的基因表達調節脂代謝。已有文獻[51]報道,在非酒精性脂肪性肝病小鼠模型中,使用雙重FXR/TGR5激動劑治療可減輕肝臟脂肪變性并抑制肝臟炎癥;雙重激動劑可抑制巨噬細胞產生促炎因子,增加IL-10的產生,并導致由肥胖誘導的M1到M2型巨噬細胞的轉換。目前,FXR激動劑奧貝膽酸用于NASH 患者的臨床試驗正在開展[52]。
5 小結與展望
隨著對肝臟中巨噬細胞極化以及免疫代謝的進一步研究,靶向巨噬細胞代謝已是肝纖維化治療及藥物開發的可行策略。靶向巨噬細胞代謝具有改善肝纖維化和較少藥物副作用的獨特優勢,但也存在一些不足:如代謝靶標的非特異性,以及還需對巨噬細胞代謝的時空特征進行準確描述,使靶向巨噬細胞代謝治療更為精確。
總的來說,近十年免疫代謝方面的重大進展表明,免疫與代謝的相互作用是包括肝纖維化在內的許多疾病的核心。不同代謝方式及代謝產物對肝纖維化過程中巨噬細胞極化產生不同影響,從而影響肝纖維化的進展與消退,靶向巨噬細胞代謝將是肝纖維化治療的新策略。
利益沖突聲明:所有作者均聲明不存在利益沖突。
作者貢獻聲明:許鈞、金衛林參與文章的結構設計;許鈞起草文章初稿;李汛、金衛林修改文章關鍵內容;所有作者閱讀并批準最終稿。
參考文獻:
[1]
ROEHLEN N, CROUCHET E, BAUMERT TF. Liver fibrosis: Mechanistic concepts and therapeutic perspectives[J]. Cells, 2020, 9(4): 875. DOI: 10.3390/cells9040875.
[2]PHAN AT, GOLDRATH AW, GLASS CK. Metabolic and epigenetic coordination of T cell and macrophage immunity[J]. Immunity, 2017, 46(5): 714-729. DOI: 10.1016/j.immuni.2017.04.016.
[3]
ONEILL LA, KISHTON RJ, RATHMELL J. A guide to immunometabolism for immunologists[J]. Nat Rev Immunol, 2016, 16(9): 553-565. DOI: 10.1038/nri.2016.70.
[4]van DER HEIDE D, WEISKIRCHEN R, BANSAL R. Therapeutic targeting of hepatic macrophages for the treatment of liver diseases[J]. Front Immunol, 2019, 10: 2852. DOI: 10.3389/fimmu.2019.02852.
[5]LI P, HE K, LI J, et al. The role of Kupffer cells in hepatic diseases[J]. Mol Immunol, 2017, 85: 222-229. DOI: 10.1016/j.molimm.2017.02.018.
[6]GUILLOT A, TACKE F. Liver macrophages: Old dogmas and new insights[J]. Hepatol Commun, 2019, 3(6): 730-743. DOI: 10.1002/hep4.1356.
[7]WANG C, MA C, GONG L, et al. Macrophage polarization and its role in liver disease[J]. Front Immunol, 2021, 12: 803037. DOI: 10.3389/fimmu.2021.803037.
[8]TACKE F. Targeting hepatic macrophages to treat liver diseases[J]. J Hepatol, 2017, 66(6): 1300-1312. DOI: 10.1016/j.jhep.2017.02.026.
[9]YUNNA C, MENGRU H, LEI W, et al. Macrophage M1/M2 polarization[J]. Eur J Pharmacol, 2020, 877: 173090. DOI: 10.1016/j.ejphar.2020.173090.
[10]WU K, MA J, ZHAN Y, et al. Down-regulation of microRNA-214 contributed to the enhanced mitochondrial transcription factor A and inhibited proliferation of colorectal cancer cells[J]. Cell Physiol Biochem, 2018, 49(2): 545-554. DOI: 10.1159/000492992.
[11]GONG J, LI J, DONG H, et al. Inhibitory effects of berberine on proinflammatory M1 macrophage polarization through interfering with the interaction between TLR4 and MyD88[J]. BMC Complement Altern Med, 2019, 19(1): 314. DOI: 10.1186/s12906-019-2710-6.
[12]WANG F, ZHANG S, JEON R, et al. Interferon gamma induces reversible metabolic reprogramming of M1 macrophages to sustain cell viability and pro-inflammatory activity[J]. EBioMedicine, 2018, 30: 303-316. DOI: 10.1016/j.ebiom.2018.02.009.
[13]SINGLA RD, WANG J, SINGLA DK. Regulation of Notch 1 signaling in THP-1 cells enhances M2 macrophage differentiation[J]. Am J Physiol Heart Circ Physiol, 2014, 307(11): H1634-H1642. DOI: 10.1152/ajpheart.00896.2013.
[14]WEI W, LI ZP, BIAN ZX, et al. Astragalus polysaccharide RAP induces macrophage phenotype polarization to M1 via the Notch signaling pathway[J]. Molecules, 2019, 24(10): 2016. DOI: 10.3390/molecules24102016.
[15]
SHENG J, ZHANG B, CHEN Y, et al. Capsaicin attenuates liver fibrosis by targeting Notch signaling to inhibit TNF-α secretion from M1 macrophages[J]. Immunopharmacol Immunotoxicol, 2020, 42(6): 556-563. DOI: 10.1080/08923973.2020.1811308.
[16]SHAPOURI-MOGHADDAM A, MOHAMMADIAN S, VAZINI H, et al. Macrophage plasticity, polarization, and function in health and disease[J]. J Cell Physiol, 2018, 233(9): 6425-6440. DOI: 10.1002/jcp.26429.
[17]GAO S, ZHOU J, LIU N, et al. Curcumin induces M2 macrophage polarization by secretion IL-4 and/or IL-13[J]. J Mol Cell Cardiol, 2015, 85: 131-139. DOI: 10.1016/j.yjmcc.2015.04.025.
[18]LU H, WU L, LIU L, et al. Quercetin ameliorates kidney injury and fibrosis by modulating M1/M2 macrophage polarization[J]. Biochem Pharmacol, 2018, 154: 203-212. DOI: 10.1016/j.bcp.2018.05.007.
[19]DOU L, SHI X, HE X, et al. Macrophage phenotype and function in liver disorder[J]. Front Immunol, 2019, 10: 3112. DOI: 10.3389/fimmu.2019.03112.
[20]CHENG D, CHAI J, WANG H, et al. Hepatic macrophages: Key players in the development and progression of liver fibrosis[J]. Liver Int, 2021, 41(10): 2279-2294. DOI: 10.1111/liv.14940.
[21]SCHWABE RF, TABAS I, PAJVANI UB. Mechanisms of fibrosis development in nonalcoholic steatohepatitis[J]. Gastroenterology, 2020, 158(7): 1913-1928. DOI: 10.1053/j.gastro.2019.11.311.
[22]TSUCHIDA T, FRIEDMAN SL. Mechanisms of hepatic stellate cell activation[J]. Nat Rev Gastroenterol Hepatol, 2017, 14(7): 397-411. DOI: 10.1038/nrgastro.2017.38.
[23]ROBERT S, GICQUEL T, VICTONI T, et al. Involvement of matrix metalloproteinases (MMPs) and inflammasome pathway in molecular mechanisms of fibrosis[J]. Biosci Rep, 2016, 36(4): e00360. DOI: 10.1042/BSR20160107.
[24]MEHTA KJ, FARNAUD SJ, SHARP PA. Iron and liver fibrosis: Mechanistic and clinical aspects[J]. World J Gastroenterol, 2019, 25(5): 521-538. DOI: 10.3748/wjg.v25.i5.521.
[25]ROTH KJ, COPPLE BL. Role of hypoxia-inducible factors in the development of liver fibrosis[J]. Cell Mol Gastroenterol Hepatol, 2015, 1(6): 589-597. DOI: 10.1016/j.jcmgh.2015.09.005.
[26]LIASKOU E, ZIMMERMANN HW, LI KK, et al. Monocyte subsets in human liver disease show distinct phenotypic and functional characteristics[J]. Hepatology, 2013, 57(1): 385-398. DOI: 10.1002/hep.26016.
[27]KISSELEVA T, BRENNER D. Molecular and cellular mechanisms of liver fibrosis and its regression[J]. Nat Rev Gastroenterol Hepatol, 2021, 18(3): 151-166. DOI: 10.1038/s41575-020-00372-7.
[28]TACKE F, ZIMMERMANN HW. Macrophage heterogeneity in liver injury and fibrosis[J]. J Hepatol, 2014, 60(5): 1090-1096. DOI: 10.1016/j.jhep.2013.12.025.
[29]
FENG M, DING J, WANG M, et al. Kupffer-derived matrix metalloproteinase-9 contributes to liver fibrosis resolution[J]. Int J Biol Sci, 2018, 14(9): 1033-1040. DOI: 10.7150/ijbs.25589.
[30]YU B, QIN SY, HU BL, et al. Resveratrol improves CCL4-induced liver fibrosis in mouse by upregulating endogenous IL-10 to reprogramme macrophages phenotype from M(LPS) to M(IL-4)[J]. Biomed Pharmacother, 2019, 117: 109110. DOI: 10.1016/j.biopha.2019.109110.
[31]VAN DEN BOSSCHE J, ONEILL LA, MENON D. Macrophage immunometabolism: Where are we (going)?[J]. Trends Immunol, 2017, 38(6): 395-406. DOI: 10.1016/j.it.2017.03.001.
[32]
CASTEGNA A, GISSI R, MENGA A, et al. Pharmacological targets of metabolism in disease: Opportunities from macrophages[J]. Pharmacol Ther, 2020, 210: 107521. DOI: 10.1016/j.pharmthera.2020.107521.
[33]FERNNDEZ-VELEDO S, CEPERUELO-MALLAFR V, VENDRELL J. Rethinking succinate: an unexpected hormone-like metabolite in energy homeostasis[J]. Trends Endocrinol Metab, 2021, 32(9): 680-692. DOI: 10.1016/j.tem.2021.06.003.
[34]BIEGHS V, WALENBERGH SM, HENDRIKX T, et al. Trapping of oxidized LDL in lysosomes of Kupffer cells is a trigger for hepatic inflammation[J]. Liver Int, 2013, 33(7): 1056-1061. DOI: 10.1111/liv.12170.
[35]BIEGHS V, WOUTERS K, VAN GORP PJ, et al. Role of scavenger receptor A and CD36 in diet-induced nonalcoholic steatohepatitis in hyperlipidemic mice[J]. Gastroenterology, 2010, 138(7): 2477-2486. DOI: 10.1053/j.gastro.2010.02.051.
[36]LEROUX A, FERRERE G, GODIE V, et al. Toxic lipids stored by Kupffer cells correlates with their pro-inflammatory phenotype at an early stage of steatohepatitis[J]. J Hepatol, 2012, 57(1): 141-149. DOI: 10.1016/j.jhep.2012.02.028.
[37]KANAMORI Y, TANAKA M, ITOH M, et al. Iron-rich Kupffer cells exhibit phenotypic changes during the development of liver fibrosis in NASH[J]. iScience, 2021, 24(2): 102032. DOI: 10.1016/j.isci.2020.102032.
[38]HANDA P, THOMAS S, MORGAN-STEVENSON V, et al. Iron alters macrophage polarization status and leads to steatohepatitis and fibrogenesis[J]. J Leukoc Biol, 2019, 105(5): 1015-1026. DOI: 10.1002/JLB.3A0318-108R.
[39]LESLIE J, MACIA MG, LULI S, et al. c-Rel orchestrates energy-dependent epithelial and macrophage reprogramming in fibrosis[J]. Nat Metab, 2020, 2(11): 1350-1367. DOI: 10.1038/s42255-020-00306-2.
[40]XU F, GUO M, HUANG W, et al. Annexin A5 regulates hepatic macrophage polarization via directly targeting PKM2 and ameliorates NASH[J]. Redox Biol, 2020, 36: 101634. DOI: 10.1016/j.redox.2020.101634.
[41]GAO YS, QIAN MY, WEI QQ, et al. WZ66, a novel acetyl-CoA carboxylase inhibitor, alleviates nonalcoholic steatohepatitis (NASH) in mice[J]. Acta Pharmacol Sin, 2020, 41(3): 336-347. DOI: 10.1038/s41401-019-0310-0.
[42]WENG SY, SCHUPPAN D. AMPK regulates macrophage polarization in adipose tissue inflammation and NASH[J]. J Hepatol, 2013, 58(3): 619-621. DOI: 10.1016/j.jhep.2012.09.031.
[43]LOOMBA R, KAYALI Z, NOUREDDIN M, et al. GS-0976 reduces hepatic steatosis and fibrosis markers in patients with nonalcoholic fatty liver disease[J]. Gastroenterology, 2018, 155(5): 1463-1473. DOI: 10.1053/j.gastro.2018.07.027.
[44]FRANCQUE S, SZABO G, ABDELMALEK MF, et al. Nonalcoholic steatohepatitis: the role of peroxisome proliferator-activated receptors[J]. Nat Rev Gastroenterol Hepatol, 2021, 18(1): 24-39. DOI: 10.1038/s41575-020-00366-5.
[45]ORFILA C, LEPERT JC, ALRIC L, et al. Immunohistochemical distribution of activated nuclear factor kappaB and peroxisome proliferator-activated receptors in carbon tetrachloride-induced chronic liver injury in rats[J]. Histochem Cell Biol, 2005, 123(6): 585-593. DOI: 10.1007/s00418-005-0785-2.
[46]STIENSTRA R, MANDARD S, TAN NS, et al. The Interleukin-1 receptor antagonist is a direct target gene of PPARalpha in liver[J]. J Hepatol, 2007, 46(5): 869-877. DOI: 10.1016/j.jhep.2006.11.019.
[47]IP E, FARRELL G, HALL P, et al. Administration of the potent PPARalpha agonist, Wy-14,643, reverses nutritional fibrosis and steatohepatitis in mice[J]. Hepatology, 2004, 39(5): 1286-1296. DOI: 10.1002/hep.20170.
[48]GILGENKRANTZ H, MALLAT A, MOREAU R, et al. Targeting cell-intrinsic metabolism for antifibrotic therapy[J]. J Hepatol, 2021, 74(6): 1442-1454. DOI: 10.1016/j.jhep.2021.02.012.
[49]
GALLI A, CRABB DW, CENI E, et al. Antidiabetic thiazolidinediones inhibit collagen synthesis and hepatic stellate cell activation in vivo and in vitro[J]. Gastroenterology, 2002, 122(7): 1924-1940. DOI: 10.1053/gast.2002.33666.
[50]
RATZIU V, CHARLOTTE F, BERNHARDT C, et al. Long-term efficacy of rosiglitazone in nonalcoholic steatohepatitis: results of the fatty liver improvement by rosiglitazone therapy (FLIRT 2) extension trial[J]. Hepatology, 2010, 51(2): 445-453. DOI: 10.1002/hep.23270.
[51]
MCMAHAN RH, WANG XX, CHENG LL, et al. Bile acid receptor activation modulates hepatic monocyte activity and improves nonalcoholic fatty liver disease[J]. J Biol Chem, 2013, 288(17): 11761-11770. DOI: 10.1074/jbc.M112.446575.
[52]
YOUNOSSI ZM, RATZIU V, LOOMBA R, et al. Obeticholic acid for the treatment of non-alcoholic steatohepatitis: interim analysis from a multicentre, randomised, placebo-controlled phase 3 trial[J]. Lancet, 2019, 394(10215): 2184-2196. DOI: 10.1016/S0140-6736(19)33041-7.
收稿日期:
2022-08-02;錄用日期:2022-09-21
本文編輯:葛俊
