Co摻雜對Ni/NiAlOx催化劑結構和菲加氫飽和性能的影響
2023-05-22 03:52:38荊潔穎李文英
煤炭學報 2023年3期
關鍵詞:催化劑
陳 羽,李 澤,荊潔穎,李文英
(太原理工大學 省部共建煤基能源清潔高效利用國家重點實驗室,山西 太原 030024)
煤炭經過高溫干餾后可得到煤焦油,通過有機結合化學、物理方法可以用于制備高附加值化學品、高性能燃料,實現煤焦油深度加工利用[1-3]。煤焦油中富含稠環芳烴,具有毒性、致癌性的稠環芳烴會對環境造成污染,而且當燃料油中芳烴含量過高時會降低油品質量;加氫飽和后的環烷烴,由于具有高密度、高熱值、高熱穩定性、燃燒性能佳等特點是航空航天燃料的理想組分[4-10]。稠環芳烴加氫飽和制環烷烴的關鍵在于研制高性能催化劑。
在稠環芳烴加氫飽和方面,催化劑活性組分主要為過渡金屬,過渡金屬與稠環芳烴間的吸附為π絡合吸附[11],該理論經常用于芳烴、烯烴分離、芳烴加氫飽和領域。當過渡金屬原子與電負性較大的原子結合、或利用金屬與載體間強相互作用力原則時,電子會朝向電負性較大的原子偏移,使得過渡金屬呈現“缺電子”狀態,當與帶有π電子的芳烴分子接觸時易于吸附在“缺電子”狀態的過渡金屬表面,形成σ鍵,過渡金屬d電子會與芳烴分子間形成反饋π鍵,σ-π鍵的共同作用強化了π絡合吸附效果[12-15]。NIU等[16]為了驗證處于“缺電子”狀態Pt有利于菲加氫反應進行,通過VASP理論計算軟件驗證。Pt處于“缺電子”狀態時,與菲分子之間的吸附能明顯高于Pt處于“富電子”狀態時,有利于菲加氫反應進行。貴金屬Pt、Pd催化劑[17]在低溫條件下具有較高的加氫活性[18],但存在著價格昂貴、地殼中含量較少且對含硫含氮化合物較為敏感的劣勢,而Ni基催化劑具有價格低廉、加氫能力強的優勢。筆者所在課題組前期以溶膠凝膠法制備鎳鋁尖晶石催化劑[19],具有較強的菲加氫飽和能力,該催化劑主要利用了活性金屬Ni與載體NiAl2O4之間的強相互作用使得活性組分Ni呈現出“缺電子”狀態,有利于芳烴的吸附與活化。該催化劑初始活性較高,隨著反應進行全氫菲選擇性呈現出下降趨勢,主要是由于在反應過程中催化劑活性組分Ni電子結構發生變化,“缺電子”程度逐漸增加,不利于芳烴的吸附。
為提高鎳鋁尖晶石催化劑的穩定性,如何優化催化劑活性組分的電子結構是關鍵。目前,調控催化劑電子結構的方法主要包含雜原子摻雜[17]、利用應力效應和配體效應[20]、制備特殊納米限域結構[21]、異質結構設計[22]。其中雜原子摻雜是最簡單、直接的調控催化劑電子結構的方法,常用于稠環芳烴加氫飽和[23-24]、電解水制氫反應[17,25-26]、二氧化碳還原[27-28]等相關領域。通過調控摻入雜原子的種類和含量可以對催化劑中電子偏移狀態[29]、活性組分價態[30]、活性組分d帶中心位置[31]進行設計,從而達到提高催化劑性能的目的[22,25,32-34]。常用金屬雜原子有Fe、Co、鑭系元素等[17]。其中,金屬Ni與Co原子序數相鄰,2者具有類似的幾何和電子結構,Co具有提高催化劑活性、增強抗積炭能力等優勢[35],經常被用于生物質轉化[36]、電催化析氫反應[33]、甲烷干重整[37]、費托合成至高級醇[38]等領域,與單一金屬Ni相比,金屬Ni與Co形成合金后可極大提高催化劑性能,2者結合可以改變單一金屬的表面性質[39],合金相形成后會進一步提升催化劑活性組分的分散度、穩定性、抗失活性[40]。AHMED等[41]通過摻雜金屬Ni、Co和Mn改性Fe/MgO催化劑,探究不同金屬摻雜對于甲烷分解、制氫產率的影響,結果表明3種金屬摻雜后均提升了Fe/MgO催化劑性能,當金屬Ni摻雜量為3%時催化劑性能最佳,適量金屬摻入更利于活性金屬分散。DEEPAK等[42]為了提升催化劑對愈創木酚的加氫脫氧能力,制備了NiCo/Al2O3催化劑,Co摻雜后提高了催化劑活性組分的分散度,有利于形成穩定性更強的Ni活性位點,而且NiCo合金形成后會產生獨特的電子特性,有利于反應物的吸附與活化[36,38,42-44]。GAO等[35]為了制備高性能、穩定性強的甲烷干重整反應催化劑,制備了NiCo/SiO2催化劑,與單金屬Ni催化劑相比,NiCo合金的形成有利于減小催化劑的晶粒尺寸,制備催化劑過程中油酸油胺的加入有利于形成高度分散、均勻的NiCo合金催化劑,均勻NiCo合金形成有利于原料的解離,減少積碳的形成,使得該催化劑具有較高的CO2、CH4轉化率和穩定性。
為穩定催化劑活性金屬Ni“缺電子”程度,本研究將通過摻雜Co對Ni/NiAlOx催化劑進行改性,調控活性組分Ni電子結構,優化催化劑穩定性。通過制備不同金屬Co摻雜量的Ni-Co/NiAlOx催化劑,探究金屬Co摻雜量對于催化劑結構及菲加氫性能的影響。
1 實 驗
1.1 Ni-Co/NiAlOx催化劑的制備
采用溶膠凝膠法制備Ni/NiAlOx催化劑[19]:依次稱取六水合硝酸鎳(Ni(NO3)2·6H2O)、九水合硝酸鋁(Al(NO3)3·9H2O)、一水合檸檬酸(C6H8O7·H2O)溶解于去離子水中,Ni/Al物質的量比為0.725∶1,去離子水∶檸檬酸∶(Ni2++Al3+)物質的量比為40∶1∶1。25 ℃條件下攪拌1 h后,在80 ℃水浴下緩慢蒸干水分得到濕凝膠,濕凝膠于室溫下放置1 h后,采用兩段式溫度(100 ℃干燥12 h,120 ℃干燥12 h)干燥。將干燥后的樣品焙燒,焙燒條件:1 ℃/min的升溫速率,由室溫升溫至650 ℃,恒溫2 h。催化劑命名為Ni/NiAlOx催化劑。
采用等體積浸漬法制備Ni-Co/NiAlOx催化劑。稱取適量Ni/NiAlOx催化劑,以六水合硝酸鈷(Co(NO3)2·6H2O)為前驅體鹽,調控金屬Co摻雜量(質量分數)分別為1%、2%、3%、4%,室溫條件下攪拌24 h緩慢蒸發水分,催化劑在120 ℃溫度下干燥12 h,再以2 ℃/min的升溫速率,從室溫升溫至400 ℃恒溫2 h焙燒。依據不同Co摻雜量分別命名為Ni-1Co/NiAlOx、Ni-2Co/NiAlOx、Ni-3Co/NiAlOx、Ni-4Co/NiAlOx。
1.2 Ni-Co/NiAlOx催化劑表征
催化劑的晶相結構使用日本理學株式會社生產的X射線衍射裝置測試,裝置中以單色Cu靶為輻射源(λ=0.154 nm)。將催化劑研磨成粉末后,制樣、測量,掃描角度區間10°~90°,掃描速率為4(°)/min。
催化劑孔徑結構、比表面積的測定在美國康塔儀器公司生產的Quantachrome Autosorb-iQ型物理吸附儀測試。采用BET、BJH方法分析樣品數據。
催化劑的還原性能(H2-TPR)、還原度及分散度在美國麥克默瑞提克公司生產的全自動程序升溫Autochem II 2920型化學吸附儀測試,采用CO脈沖對催化劑活性組分分散度進行測試,測試條件:稱取80 mg左右催化劑,在520 ℃條件下,氫氣氣氛下原位還原5 h,降溫至50 ℃后,采用CO連續脈沖吸附20次。
催化劑形貌、粒徑以及晶格條紋在日本電子株式會社生產的高分辨透射電子顯微鏡JEM-2100F測試。
催化劑的X射線吸收近邊吸收光譜(XANES)在新加坡同步輻射光源中心(SSLS)測試。通過XANES測試活性組分Nid帶電子得失數,即d電荷密度,計算公式[45]為
(1)
(2)
其中,Δn為d電荷密度,e;Asample為催化劑L3邊或L2邊吸收峰峰面積;ANi/Al2O3為Ni/Al2O3催化劑L3邊或L2邊吸收峰峰面積;σ/n3d(Ni)為Ni 3d軌道每摩爾空位的吸收截面;σ(Ni)為Ni元素L2,3邊峰面積;σ(Cu)為Cu元素L2,3邊峰面積;n3d(Ni)為Ni 3d電子數;n3d(Cu)為Cu 3d電子數,參照文獻中相關參數[46-49],計算得σ/n3d(Ni)為13.73。
1.3 催化劑活性評價
采用固定床反應器評價催化劑性能,催化劑用量為100 mg。反應前催化劑需先進行原位還原。催化劑還原條件為:以3 ℃/min的升溫速率由室溫升溫至520 ℃,還原5 h,氫氣流量為50 mL/min。反應條件為:溫度300 ℃,壓力為5.0 MPa,原料為含有1%菲/十氫萘溶劑或1%對稱八氫菲/十氫萘溶劑,進料速率為6 mL/h,氫氣流量為60 mL/min,氫油體積比為600,重時空速為52 h-1,每1 h接取產物進行分析。產物分析采用島津GC-2010氣相色譜,以十四烷作為內標對產物進行定量分析。
菲或對稱八氫菲轉化率公式為
(3)
式中,X為菲或對稱八氫菲轉化率,%;Nin為原料菲或對稱八氫菲物質的量,mmol;Nout為產物中菲或對稱八氫菲物質的量,mmol。
菲加氫中間產物及加氫飽和產物選擇性公式為
(4)
式中,Si為菲加氫中間產物及加氫飽和產物選擇性,%;Ni為菲加氫中間產物及加氫飽和產物物質的量,mmol;下角標i為不同的中間產物(二氫菲、四氫菲、不對稱八氫菲、對稱八氫菲、全氫菲);ΣNi為所有加氫產物物質的量,mmol。
菲或對稱八氫菲轉化量公式為
(5)
式中,Z為菲或對稱八氫菲轉化量(單位時間單位質量活性組分上原料菲或對稱八氫菲的轉化量),mmol/(g·h);m為催化劑質量,g;y為催化劑中活性組分質量分數,%;r為催化劑還原度,%。
在排除內外擴散的前提下,提高重時空速,將原料轉化率控制在20%以下,采用轉化頻率探究催化劑的本征活性。轉化頻率計算公式為
(6)
式中,TOF為轉化頻率,s-1;F為原料菲摩爾流量,mmol/s;M為催化劑中活性組分Ni和Co物質的量,mmol;f為活性組分分散度,%,采用CO脈沖測得。
2 結果與討論
2.1 不同Co負載量催化劑的結構表征
2.1.1 晶相結構
采用XRD分析催化劑晶相結構,探究Co摻雜量對Ni/NiAlOx催化劑晶相結構的影響。圖1為不同Co摻雜量催化劑焙燒后的XRD譜圖。5種催化劑主要包含3處衍射峰,分別對應NiAl2O4(PDF#78-1601)的(311)、(400)、(440)晶面。鎳鋁尖晶石具有一定的特殊性,會呈現出部分倒置現象,四面體位點會優先被Al3+占據,部分Ni2+與Al3+占據八面體位點,八面體位點的Ni2+相比于四面體位點的Ni2+更容易被還原成單質Ni,而且從鎳鋁尖晶石催化劑中被還原出的金屬Ni粒徑更小、分散程度更高[50-55]。進一步分析焙燒后催化劑晶相結構,31.4°和63.7°處的衍射峰分別對應鎳鋁尖晶石四面體位點和八面體位點,基本上未觀察到四面體位點衍射峰,而八面體位點衍射峰較強,證明Ni2+主要位于八面體位點[15]。摻雜金屬Co后催化劑晶相結構基本與未摻雜時一致,證明Co摻雜后沒有改變Ni2+主要占據八面體位點的NiAl2O4結構,金屬Co摻雜量較少,沒有觀察到氧化鈷的衍射峰。
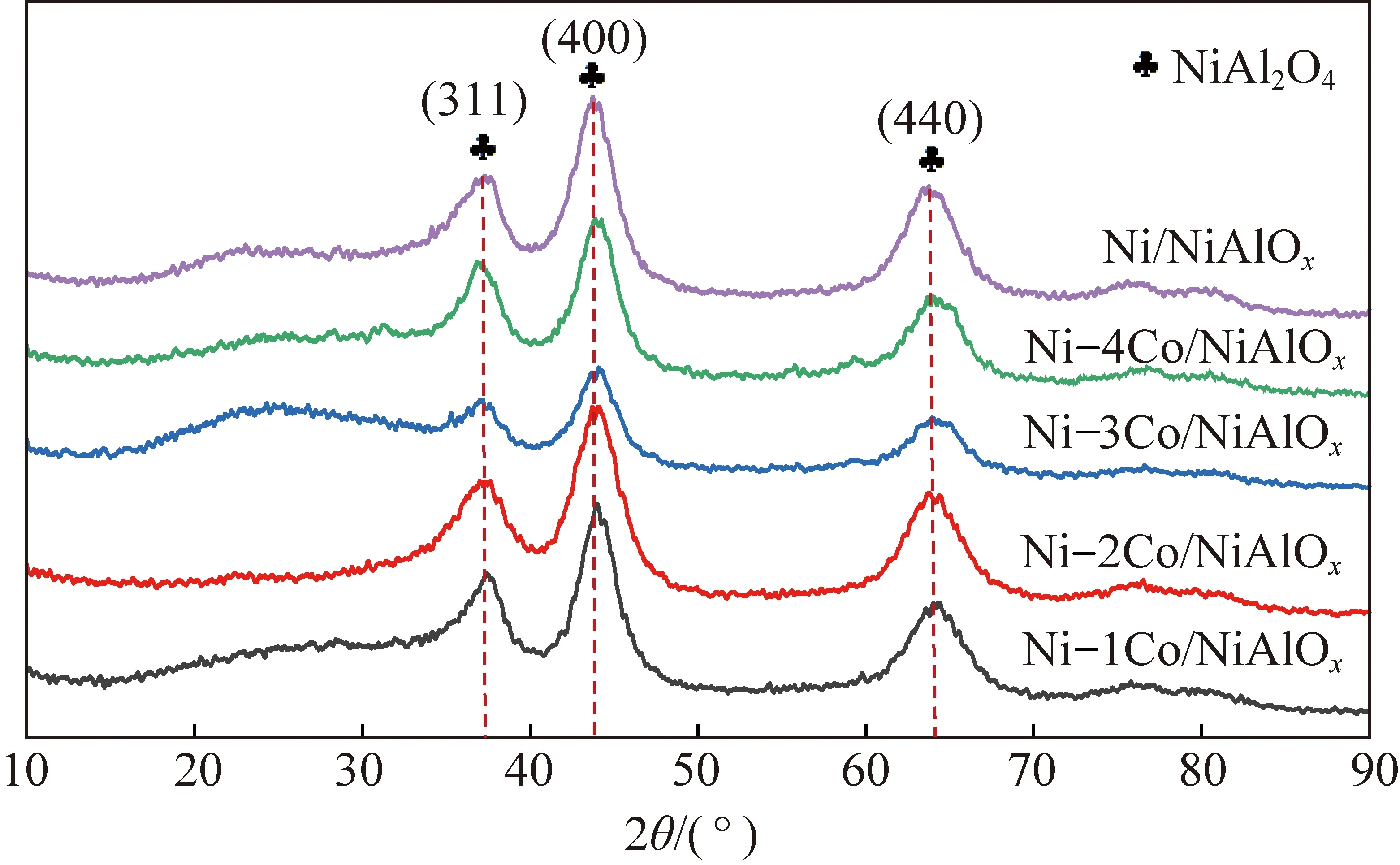
圖1 不同Co摻雜量催化劑焙燒后XRD圖Fig.1 XRD figure after calcination of catalysts with different Co doping content
圖2(a)為不同Co摻雜量催化劑還原后的XRD譜圖,可以看到:Ni/NiAlOx催化劑在44.5°、51.8°、76.3°有3處衍射峰,對應Ni(PDF#87-0712)的(111)、(200)、(220)晶面。金屬Ni和Co具有相似的原子半徑、晶格常數以及相似的面心立方晶體結構[56],Co原子容易溶解到Ni晶格中,形成NiCo合金相,Co溶解至Ni晶格中可以直接干擾Ni的電子結構,優化催化劑性能[57]。Ni衍射峰與Co(PDF#89-7903)衍射峰位置接近,不同Co摻雜量催化劑與Ni/NiAlOx催化劑具有類似的XRD衍射峰。對2θ=40°~50°時衍射峰進行局部放大研究,結果如圖2(b)所示,摻雜Co后催化劑衍射峰向低角度偏移,而且摻雜Co后催化劑衍射峰峰值均介于Ni(111)晶面和Co(111)晶面之間[35-36,58],可能形成了NiCo合金。NiCo合金主要是由于在還原過程中Co3+半徑(0.063 nm)小于Ni2+(0.069 nm),還原過程中Co溶入并取代Ni,使得Ni晶格發生不同程度的收縮[56-57,59],形成NiCo合金。以51.8°衍射峰采用謝樂公式估算催化劑活性組分粒徑,粒徑為2.5~4.3 nm。
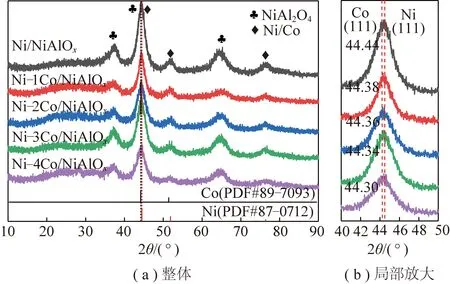
圖2 不同Co摻雜量的催化劑還原后XRD圖Fig.2 XRD figure of catalysts with different Co doping content after reduction
2.1.2 孔道結構分析
采用氮氣等溫吸脫附測試不同Co摻雜量還原后催化劑的孔徑與比表面積,測試結果見表1。與Ni/NiAlOx催化劑相比,Co摻雜后催化劑隨著Co摻雜量的增加,催化劑比表面積呈現先增大后減小的趨勢,當Co摻雜量為2%時,Ni-2Co/NiAlOx催化劑比表面積為186.2 m2/g,較大的比表面積有利于活性組分的分散。Co摻雜后催化劑的平均孔徑與總孔容均增大,平均孔徑為3.82~4.32 nm,總孔容為0.19~0.28 cm3/g,表明Co摻雜并未對催化劑的結構產生較大的影響。當Co摻雜量為1%、2%、3%時催化劑的平均孔徑大小基本一致為4.31 nm。能為菲分子提供適宜的加氫環境,有利于菲加氫飽和制備全氫菲。
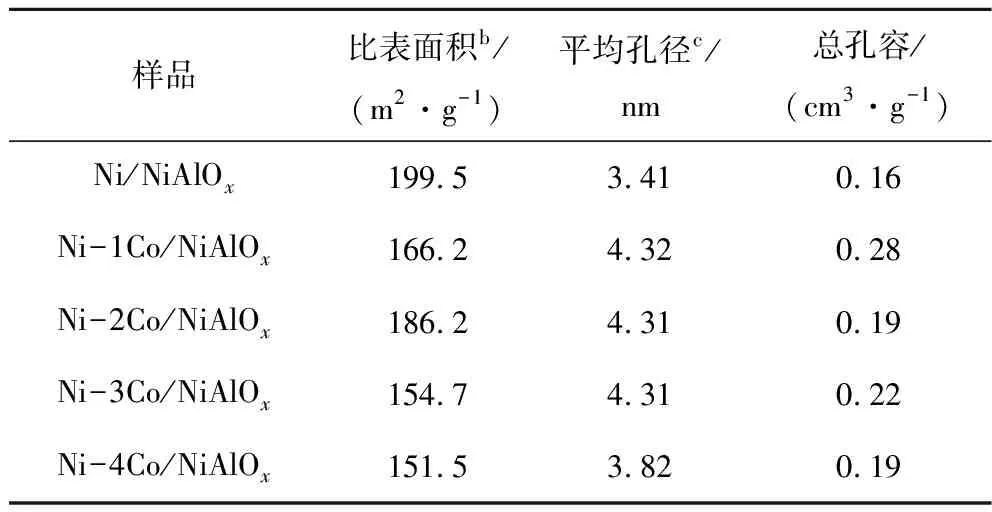
表1 不同Co摻雜量催化劑孔徑及比表面積(還原后a)
2.1.3 催化劑還原性能
采用化學吸附儀測試不同Co摻雜量催化劑還原性能。圖3為不同Co摻雜量催化劑的H2-TPR圖。其中實線代表焙燒后催化劑還原峰,虛線代表著在520 ℃下還原5 h后催化劑還原峰。Ni/NiAlOx催化劑與不同Co摻雜量催化劑均含有一個還原峰,為氧化鎳、氧化鈷、鎳鋁尖晶石中氧化鎳的還原峰[15]。Ni/NiAlOx催化劑的還原峰在655 ℃時還原峰對稱性最好,Co摻雜后催化劑還原峰朝向低溫方向偏移,還原峰面積大小側面反映了氫耗量,Co摻雜后催化劑還原峰面積增大,耗氫量增大,證明更多活性組分被還原,Co摻雜可以降低催化劑還原難度,促進活性組分還原[55]。
分別對催化劑(H2-TPR)還原峰面積以及原位還原后催化劑(H2-TPR)還原峰面積進行積分,用CuO進行標定,計算Ni/NiAlOx催化劑與不同Co摻雜量催化劑的還原度,結果見表2。Ni/NiAlOx催化劑還原度為72.4%,隨著Co摻雜量增加,催化劑還原度呈現出先增大后減小的趨勢,Co摻雜量為1%、3%時,2種催化劑還原度較為接近,分別為73.8%和73.5%,Co摻雜量為2%時,催化劑還原度提高6%,還原度為78.4%,Co摻雜量為4%時,催化劑還原度降低5.6%。與Ni/NiAlOx催化劑相比,雙金屬Ni-Co/NiAlOx催化劑,Co摻雜量為1%、2%、3%時,催化劑還原度均增大,說明適量Co摻雜可以降低催化劑還原難度,提高催化劑的還原能力,促進了活性組分的還原[60]。
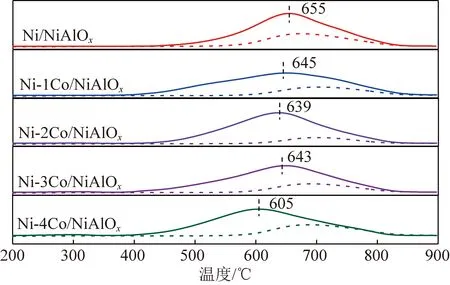
圖3 不同Co摻雜量催化劑還原前后H2-TPR圖Fig.3 H2-TPR figure before and after reduction of catalysts with different Co doping amount

表2 不同Co摻雜量催化劑還原度
2.2 催化劑加氫性能評價及構效關系
2.2.1 菲及對稱八氫菲加氫活性
在壓力5.0 MPa、反應溫度300 ℃、反應原料1%菲/十氫萘溶劑、進料速率為6 mL/h、氫氣流量為60 mL/min、氫油體積比為600、重時空速為52 h-1條件下進行6 h活性評價,評估Ni/NiAlOx催化劑和不同Co摻雜量催化劑的加氫性能,結果如圖4所示。為了排除催化劑活性組分含量對于原料菲轉化影響,采用菲轉化量描述催化劑的性能(菲轉化量計算公式見式(3))。6 h內,Ni/NiAlOx催化劑的菲轉化量(單位時間單位質量活性組分上原料菲的轉化量)基本保持不變為9.95 mmol/(g·h),全氫菲選擇性由81.9%下降至10.7%。
對比于Ni/NiAlOx催化劑,適量Co摻雜后,催化劑的活性和穩定性均得到了提升。摻雜Co后催化劑菲轉化量在9.04 ~ 10.10 mmol/(g·h),Ni/NiAlOx催化劑與不同Co摻雜量催化劑均具有較好的菲轉化能力,這與催化劑結構密不可分,5種催化劑的晶相結構中均顯示出Ni2+主要占據易于被還原出的八面體位點且通過謝樂公式計算活性組分粒徑為2.5~4.3 nm(圖2(a)),可以提供更多的活性位點,而且催化劑的平均孔徑平均孔徑為3.82~4.32 nm(表1),均大于菲分子的動力學直徑0.7 nm[16],有利于分子的擴散,所以幾種催化劑均表現出優異的菲加氫能力。與未改性催化劑相比,Ni-1Co/NiAlOx、Ni-2Co/NiAlOx、Ni-3Co/NiAlOx催化劑全氫菲初始選擇性均增大,Ni-3Co/NiAlOx催化劑全氫初始菲選擇性最高為90.8%;反應第2小時,Ni-2Co/NiAlOx催化劑全氫菲選擇性(91%)明顯高于其他幾種催化劑。由于菲加氫飽和至全氫菲是一個較為復雜的反應,在2.1.3節中通過H2-TPR表征并計算出不同Co摻雜量催化劑的還原度(表2),當Co摻雜量為1%、2%、3%時,3種催化劑還原度均高于Ni/NiAlOx催化劑(72.4%),可以提供更多的活性位點,有利于菲加氫飽和至全氫菲。隨著反應進行,幾種催化劑全氫菲選擇性呈現出不同程度下降趨勢,Ni-2Co/NiAlOx催化劑全氫菲選擇性下降趨勢緩慢,在反應第6小時,全氫菲選擇性高出未改性催化劑43%。與Co摻雜量為1%、3%、4%催化劑相比,2%為最佳Co摻雜量。
由于空間位阻的影響,對稱八氫菲是菲加氫至全氫菲的速控步驟,為了探究Ni-2Co/NiAlOx催化劑對速控步驟的影響。以對稱八氫菲為模型化合物,進行加氫性能評價。評價條件如下所示:反應溫度為300 ℃,反應壓力為5.0 MPa,氫油比600,原料為1%對稱八氫菲/十氫萘,進料速率為6 mL/h,重時空速52、104和520 h-1,固定床反應器,結果如圖5所示。隨著重時空速增大,對稱八氫菲轉化量逐漸增大,全氫菲選擇性逐漸減小。菲加氫反應是可逆的,反應過程中會存在加氫、脫氫、異構過程,產物包括全氫菲、不對稱八氫菲、四氫菲、二氫菲、菲,在52、104、520 h-1重時空速條件下,對稱八氫菲主要加氫生成全氫菲,對稱八氫菲也發生脫氫反應至菲,但菲的選擇性均小于1.0%。當重時空速為52 h-1時,全氫菲選擇性較高,為94.25%,脫氫及異構產物較少,進一步對比重時空速為52、104 h-1時催化劑對稱八氫菲加氫反應情況,對稱八氫菲轉化量分別為8.76、16.77 mmol/(g·h),全氫菲選擇性僅降低20%左右,進一步證明該催化劑有利于對稱八氫菲加氫至全氫菲,有利于菲加氫至全氫菲的速控步驟進行。
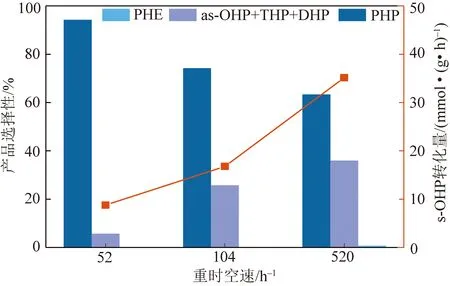
圖5 Ni-2Co/NiAlOx催化劑不同重時空速條件下 對稱八氫菲加氫性能評價Fig.5 Performance evaluation figure of symmetric octahydro phenanthrene hydrogenation over Ni-2Co/NiAlOx catalyst at different weight hourly space velocity
2.2.2 構效關系分析
因催化劑的電子結構、粒徑等均會影響催化劑的活性與穩定性。為了探究Co摻雜對未改性催化劑活性組分Ni電子結構的影響,通過X射線吸收近邊結構(XANES)表征Ni/NiAlOx催化劑和Ni-2Co/NiAlOx催化劑的電子結構,XANES光譜如圖5所示。該圖譜代表Ni 2p電子躍遷到空的3d軌道,主要顯示出2個躍遷吸收峰,NiL3邊吸收峰范圍在852~857 eV,代表Ni 2p3/2軌道躍遷至Ni 3d軌道,NiL2邊吸收峰范圍在869~873 eV間,代表Ni 2p1/2軌道躍遷至Ni 3d軌道[61-63]。與Ni-foil相比,Ni-2Co/NiAlOx催化劑的NiL3邊吸收峰強度均降低,可能是由于Co摻雜填充了Ni的d帶。
基于圖6中Ni/NiAlOx、Ni-2Co/NiAlOx與Ni/Al2O3之間的L3邊吸收峰面積之差,以及Ni/NiAlOx、Ni-2Co/NiAlOx與Ni/Al2O3之間的L2邊吸收峰面積之差計算Ni的3d軌道的d電荷密度(電子得失情況)[64],采用式(1)計算d電荷密度,Ni/NiAlOx催化劑與Ni-2Co/NiAlOx催化劑的d電荷密度分別為0.30 e和0.14 e,芳烴與過渡金屬間吸附遵循著π絡合吸附原則,活性組分處于適宜的“缺電子”狀態時有利于菲加氫反應進行。上述結果證明Co的摻雜降低并穩定活性組分Ni的“缺電子”程度,有利于芳烴與過渡金屬間的吸附與活化,提升催化劑穩定性。
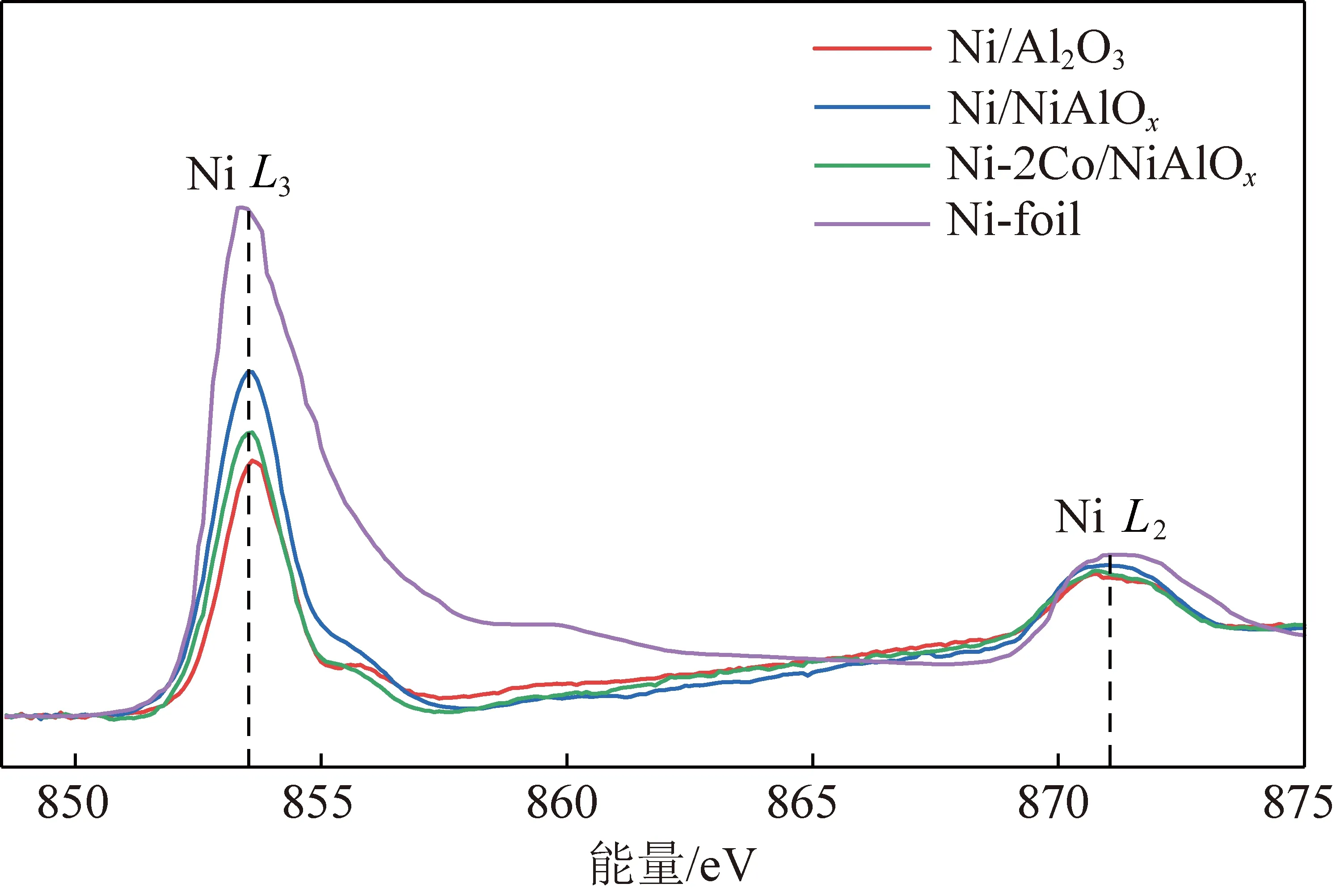
圖6 還原后的Ni/NiAlOx與Ni-2Co/NiAlOx催化劑Ni L2,3 邊X射線近邊吸收光譜Fig.6 Ni/NiAlOx catalyst after reduction and Ni-2Co/NiAlOx catalyst after reduction Ni L2,3edge XANES figure
另一方面,催化劑性能與催化劑活性組分粒徑有關[65-66]。為了獲得Ni/NiAlOx、Ni-2Co/NiAlOx催化劑形貌,對還原后和反應后催化劑進行TEM表征。圖7為Ni、Co、Al、O四種元素的X射線能譜分析,可以看出Ni-2Co/NiAlOx催化劑中Ni、Co分散均勻。通過測量晶格間距辨別出催化劑中含有Ni(111)、Ni(200)、NiAl2O4(400)、NiCo合金(111)晶面,進一步通過TEM證實NiCo合金形成,如圖8所示。對比反應前后催化劑可知,活性組分Ni顆粒并未發生團聚現象,而且Co摻雜后催化劑粒徑減小。
2.3 催化劑本征活性評價
在排除內外擴散的條件下,通過調整催化劑的質量或改變進料速率,提高空速,在菲轉化率低于20%時,對Ni/NiAlOx與Ni-2Co/NiAlOx催化劑的本征加氫活性進行評價,結果見表3。對比2種催化劑在高空速下菲轉化率,Ni/NiAlOx催化劑在1 040 h-1重時空速條件下,菲轉化率為17.6%,Ni-2Co/NiAlOx催化劑在更加嚴苛空速(1 733 h-1)條件下,菲轉化率為18.0%,高于未改性催化劑,而且Ni-2Co/NiAlOx催化劑的TOF為131.8×10-3s-1,Ni/NiAlOx催化劑的TOF為84.7×10-3s-1,Ni-2Co/NiAlOx催化劑本征活性明顯高于Ni/NiAlOx催化劑,證明Ni-2Co/NiAlOx催化劑單個活性位點具有更強的菲加氫性能。此外,Ni/NiAlOx與Ni-2Co/NiAlOx催化劑的分散度和金屬表面積基本一致,排除分散度對于加氫性能的影響,最終歸因于適量Co摻雜可以優化活性組分的電子結構,降低Ni“缺電子”程度,有利于芳烴與過渡金屬間的吸附與活化,提升催化劑性能。
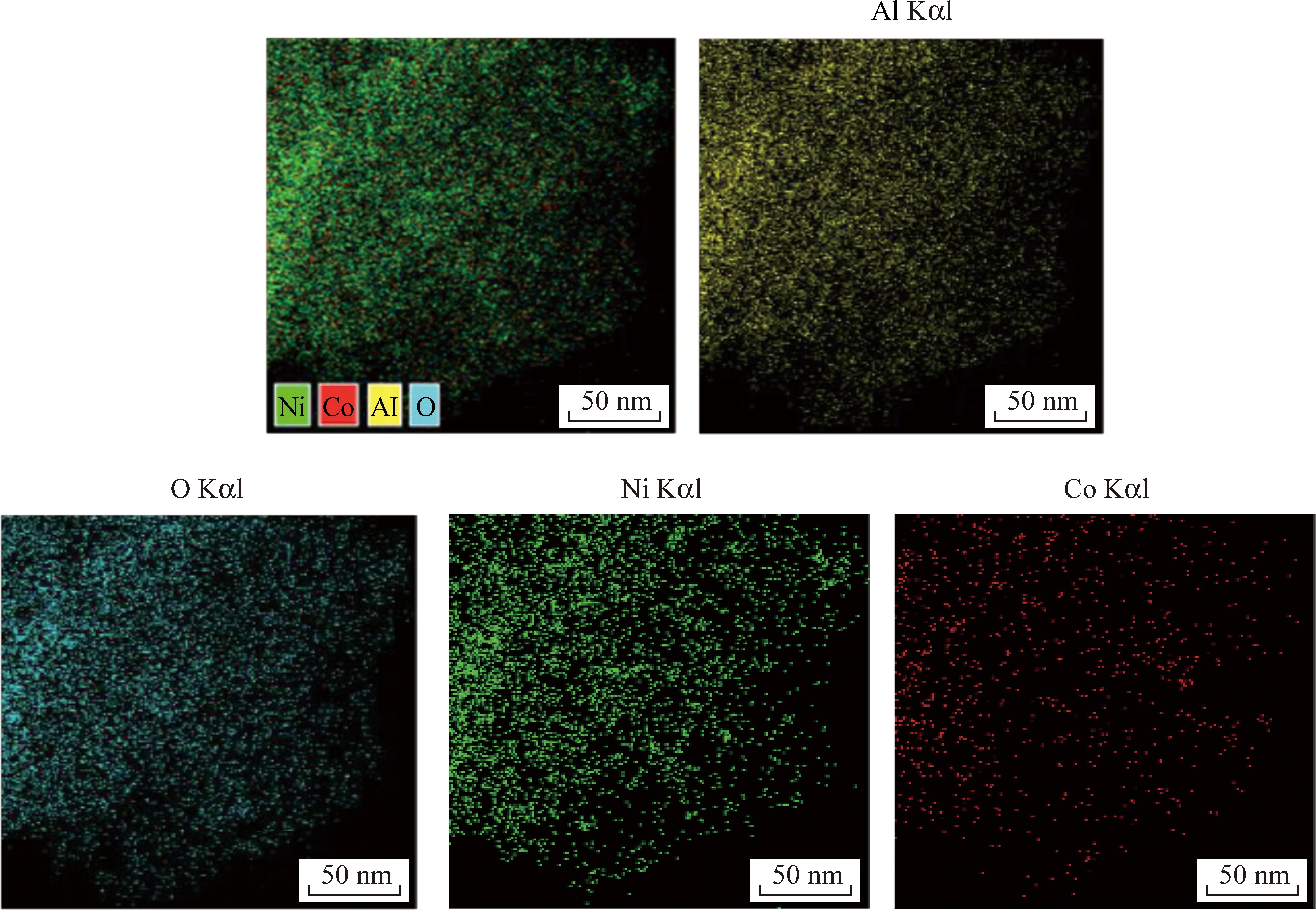
圖7 Ni-2Co/NiAlOx催化劑還原后TEM的X射線能譜分析圖(EDS)Fig.7 TEM X-ray energy spectrum analysis figure (EDS) after reduction of Ni-2Co/NiAlOx catalyst
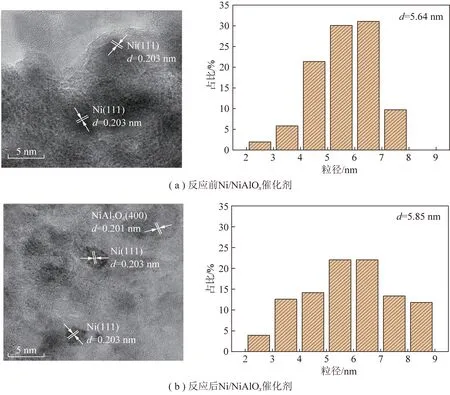
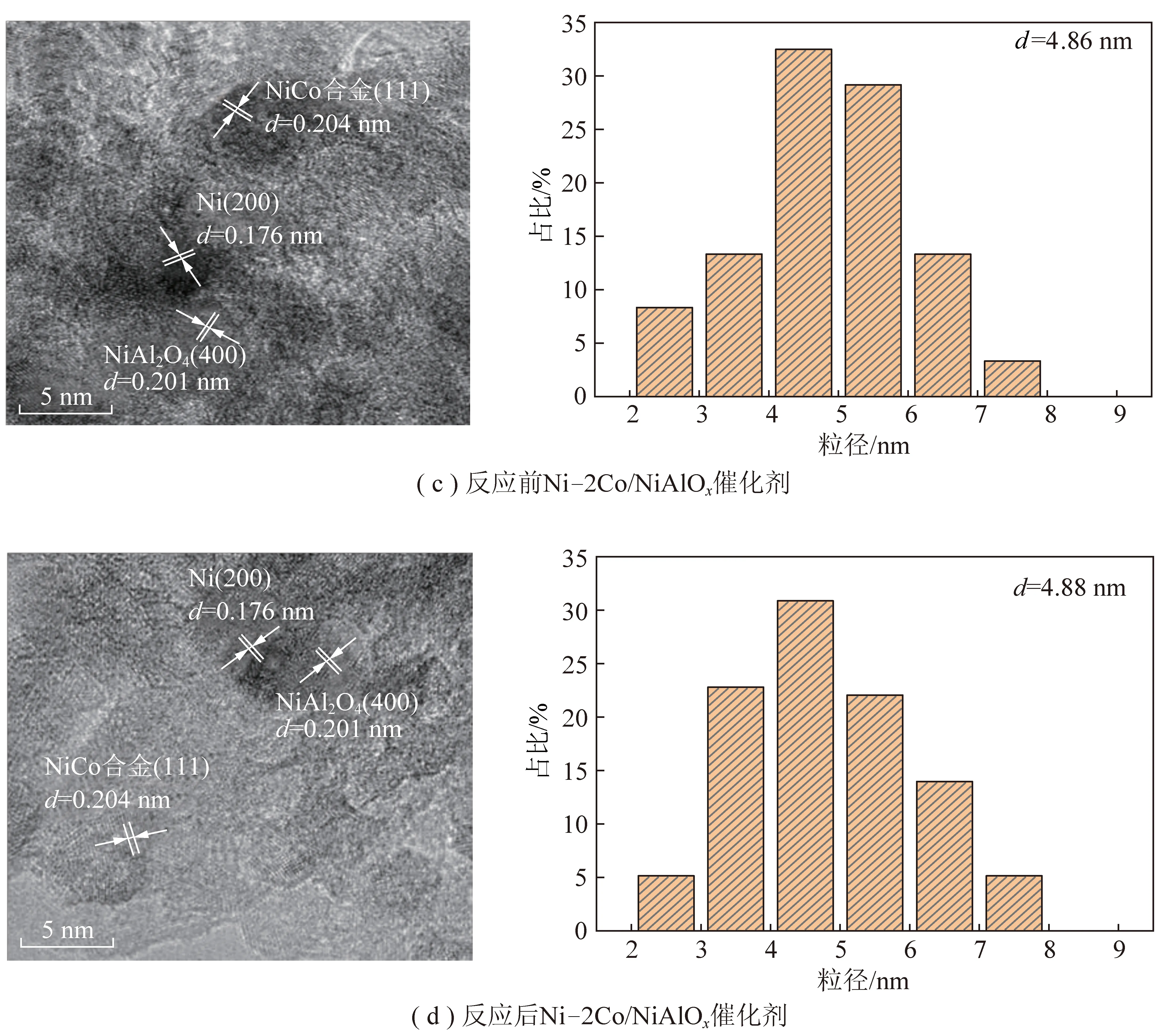
圖8 Ni/NiAlOx、Ni-2Co/NiAlOx反應前后催化劑TEM圖Fig.8 TEM figure of Ni/NiAlOx,Ni-2Co/NiAlOx catalyst before and after reaction

表3 Ni/NiAlOx與Ni-2Co/NiAlOx催化劑分散度及本征動力學
3 結 論
(1)為提升Ni/NiAlOx催化劑穩定性,選擇金屬Co作為雜原子摻雜劑,通過調控Co摻雜量優化催化劑性能。主要利用了在催化劑還原過程中,Co容易溶解入Ni晶格中形成NiCo合金,有利于活性組分還原,減小催化劑粒徑,優化活性組分電子結構。
(2)在反應壓力5.0 MPa、反應溫度300 ℃、重時空速為52 h-1條件下,評價未改性催化劑與Co摻雜量為1%、2%、3%、4%催化劑菲加氫性能,所有催化劑均表現出較高的初始全氫菲選擇性,但隨著反應進行幾種催化劑全氫菲選擇性均呈現出下降趨勢,Co摻雜量為2%時催化劑全氫菲選擇性下降趨勢最緩慢,在反應第6小時,全氫菲選擇性高出未改性催化劑全氫菲選擇性43%。此外,Ni-2Co/NiAlOx催化劑(131.8×10-3s-1)的TOF高于Ni/NiAlOx(84.7×10-3s-1)。
(3)通過XRD與TEM表征技術,證明了Ni-Co/NiAlOx催化劑形成NiCo合金相,減小催化劑粒徑;進一步通過H2-TPR計算催化劑還原度,當Co摻雜量為2%時催化劑還原度最高,為78.4%,對比于其他催化劑可以提供較多的活性位點;結合X射線近邊吸收光譜表征技術與d電荷密度計算,證明了Co摻雜優化了活性組分Ni電子結構,降低了活性組分Ni“缺電子”程度,有利于芳烴吸附與活化,提升催化劑性能。
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
