靈芝總三萜低溫提取工藝優化及其降血糖活性研究
2023-06-20 02:53:58何榮軍孫江亭孫培龍
浙江工業大學學報 2023年3期
關鍵詞:實驗
何榮軍,孫江亭,賀 丹,孫培龍
(浙江工業大學 食品科學與工程學院,浙江 杭州 310014)
靈芝(Ganodermalucidum),又稱瑞草、林中靈、瓊珍,是多孔菌科真菌靈芝的子實體[1]。《本草綱目》等書籍對靈芝有“補氣、止咳”等功能的記載[2]。多糖類和三萜類化合物是靈芝中重要的活性成分,《中國藥典》中以靈芝多糖的質量分數作為評價靈芝質量的標準,而在日本則以三萜酸的質量分數評價靈芝質量[3]。靈芝三萜類結構的復雜性和種類的多樣性決定了其藥理活性和生物活性的多樣性,具有抗腫瘤活性[1]、抗炎活性[4-5]、抗氧化活性[6-7]、降血糖活性[8]和保肝活性[9-10]等特點。靈芝三萜的傳統提取方法是乙醇熱回流法[11],近年來興起的超聲輔助提取[12-13]和超臨界CO2萃取[14-15]等工藝很大程度上解決了傳統提取時間過長等問題,但仍在較高溫度下進行提取,不利于三萜的活性保持。相較于其他提取方法,低溫高速剪切提取能夠在較低溫度下實現對活性成分的提取,既有利于活性的保持,又降低了能源的消耗,在節能減排方面具有重大意義。筆者基于單因素實驗響應面優化靈芝三萜的低溫高速剪切提取工藝,并比較了體外降血糖活性的差異,為低溫高速剪切技術在活性成分提取領域的應用提供依據。
1 材料與方法
1.1 原 料
赤芝子實體顆粒,浙江科達生物科技有限公司贈送。
1.2 試劑與儀器
香草醛(AR,99.0%)、α-葡萄糖苷酶(BR,50 U/mg)、對硝基苯基-α-D-吡喃葡萄糖苷(BR,99%),麥克林生物科技有限公司;無水乙醇(AR),上海凌峰化學試劑有限公司;熊果酸標準品(AR,>98.0%)、阿卡波糖(BR,95%),上海源葉生物科技有限公司;高氯酸(AR)、石油醚(AR,60~90 ℃)、三氯甲烷(AR),國藥集團化學試劑有限公司;碳酸氫鈉(AR,≥99.8%)、冰乙酸(AR,99.5%)、磷酸二氫鈉(AR,99.0%),羅恩化學有限公司;磷酸氫二鈉(AR,98.0%),上海畢得醫藥科技股份有限公司;鹽酸(AR),上海泰坦科技股份有限公司。
T18Digtal高速剪切機,德國儀器有限公司;752N紫外分光光度計,INESA有限公司;CR21N臺式離心機,日本日立株式會社;R-1001VN旋轉蒸發儀,鄭州長城科工貿有限公司;ALpHA2-4LD冷凍干燥機,德國Christ公司;DG5033A酶聯免疫測定儀,華東電子醫療裝備有限公司。
1.3 實驗方法
1.3.1 低溫高速剪切提取靈芝總三萜工藝流程
稱取5.0 g經中藥粉碎機粉碎的靈芝粉末,過80目篩,按一定的料液比加入乙醇攪拌均勻,用高速剪切機以12 000 r/min的轉速剪切,結束后將溶液轉移至離心瓶以4 510×g的轉速離心15 min,將上清液進行抽濾,殘渣按上述步驟繼續剪切提取,重復提取3次,合并提取液,于37 ℃下旋轉蒸發濃縮后冷凍干燥即得靈芝總三萜粗提物。提取靈芝總三萜的工藝流程如圖1所示。
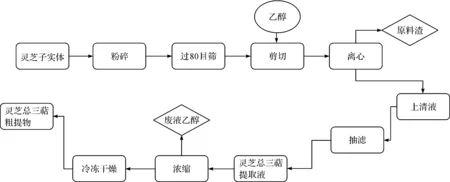
圖1 低溫高速剪切提取靈芝總三萜工藝流程
1.3.2 靈芝總三萜得率的測定
參考江紹琳等[16]的方法并稍作修改,采用熊果酸作為標準品,用無水乙醇準確配制質量濃度為0.20 mg/mL的熊果酸標準品溶液,精確量取此標準品溶液0.00,0.10,0.15,0.20,0.25,0.30,0.35,0.40 mL分別置于10 mL試管中,用無水乙醇分別補充至1.00 mL,隨后于100 ℃水浴上加熱蒸干。每管中分別加入0.40 mL 5%的香草醛-冰乙酸溶液和1.00 mL高氯酸溶液后,于60 ℃水浴中加熱15 min,冷卻后再加入5.00 mL冰乙酸,搖勻后室溫靜置15 min,用紫外-可見分光光度計在550 nm波長下測定其吸光度,建立標準曲線以測定靈芝總三萜質量分數。按照圖1得到的靈芝總三萜提取液用乙醇稀釋到一定質量濃度后,按上述方法測定總三萜得率。計算式為
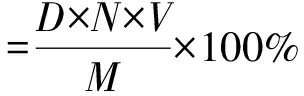
(1)
式中:D為根據標準曲線計算所得總三萜的質量濃度,μg/mL;N為稀釋倍數;V為提取液體積,mL;M為靈芝質量,g。
1.3.3 單因素實驗
基于前期預實驗結果,提取時間、料液比和乙醇體積分數是最重要的影響因素,因此重點對這3個因素進行優化。精確稱取過80目篩的靈芝子實體粉末5.0 g,按照料液比1∶25 g/mL與體積分數為95%的乙醇均勻混合,利用高速剪切技術提取2,4,6,8,10 min,主要考察提取時間對靈芝總三萜得率的影響。靈芝子實體粉末與體積分數為95%的乙醇按一定料液比均勻混合后提取6 min,主要考察料液比1∶20,1∶25,1∶30,1∶35,1∶40 g/mL對靈芝總三萜得率的影響。靈芝子實體粉末按照料液比1∶35 g/mL與體積分數為70%,75%,80%,85%,90%的乙醇分別混合后提取6 min,主要考察乙醇體積分數對靈芝總三萜得率的影響。
1.3.4 響應面設計實驗
利用Box-Behnken原理設計實驗,實驗設計中的水平及編碼如表1所示。采用Design-Expert V 10.0軟件分析所得實驗數據,優化低溫高速剪切提取靈芝總三萜的工藝參數。

表1 響應面分析實驗因素水平表
1.3.5 靈芝三萜降血糖活性分析
參考郝瑞霞[17]的方法采用堿溶酸沉法將GLEC分成酸性三萜(CA)和中性三萜(CN),按照南婷婷[11]優化后的乙醇熱回流法提取靈芝總三萜(GLEH),同樣將GLEH分成酸性三萜(HA)和中性三萜(HN),評價并比較以上樣品的α-葡萄糖苷酶活性的抑制率。參考Pinto等[18]的方法并稍作修改。用純水配制0.1 moL/L pH 6.9的PBS溶液、5 mmoL/L對硝基苯基-α-D-吡喃葡萄糖苷(pNPG)溶液和0.67 moL/L碳酸鈉終止溶液,再用PBS溶液配制0.5 U/mL的α-葡萄糖苷酶溶液。用PBS溶液配制相應的樣品溶液,以阿卡波糖為陽性對照。具體的測定體系組分和順序如表2所示。
按照表2的順序于96孔板中添加試劑并操作。加入終止液后于405 nm處測定吸光度A。計算式為
α-葡萄糖苷酶抑制率(%)=

(2)
式中:下標為組名,樣品組包括陽性對照組。進而計算每個樣品的IC50值。
2 結果與分析
2.1 靈芝總三萜質量分數標準曲線
以樣品質量濃度X為橫坐標,以測定的吸光度Y為縱坐標繪制總三萜標準曲線,具體如圖2所示。所得標準品線性回歸方程為Y=0.009 8X-0.067 9(R2=0.997 9)。
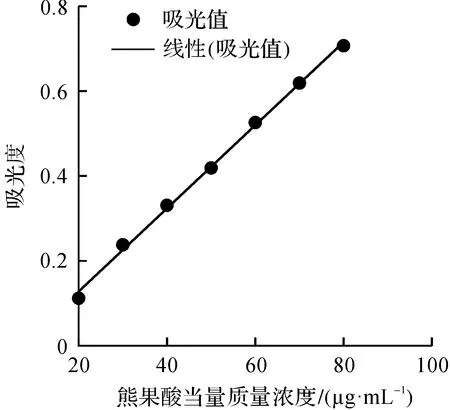
圖2 總三萜標準曲線
2.2 單因素實驗結果
2.2.1 提取時間對靈芝總三萜得率的影響
在料液比為1∶25 g/mL,乙醇體積分數為95%的實驗條件下,考察提取時間2,4,6,8,10 min對靈芝總三萜得率的影響規律,結果如圖3所示。隨著提取時間增加,靈芝總三萜得率呈先上升后下降的趨勢,提取時間為2~6 min,靈芝總三萜溶出量逐漸增加,在提取時間為6 min時得率達到最大值,此后再增加提取時間,靈芝總三萜的得率略有下降后趨于穩定,得率略有下降,其原因可能是隨著提取時間的增加,溶液逐漸達到飽和狀態,進而有一定程度析出,因此提取時間選擇為6 min。
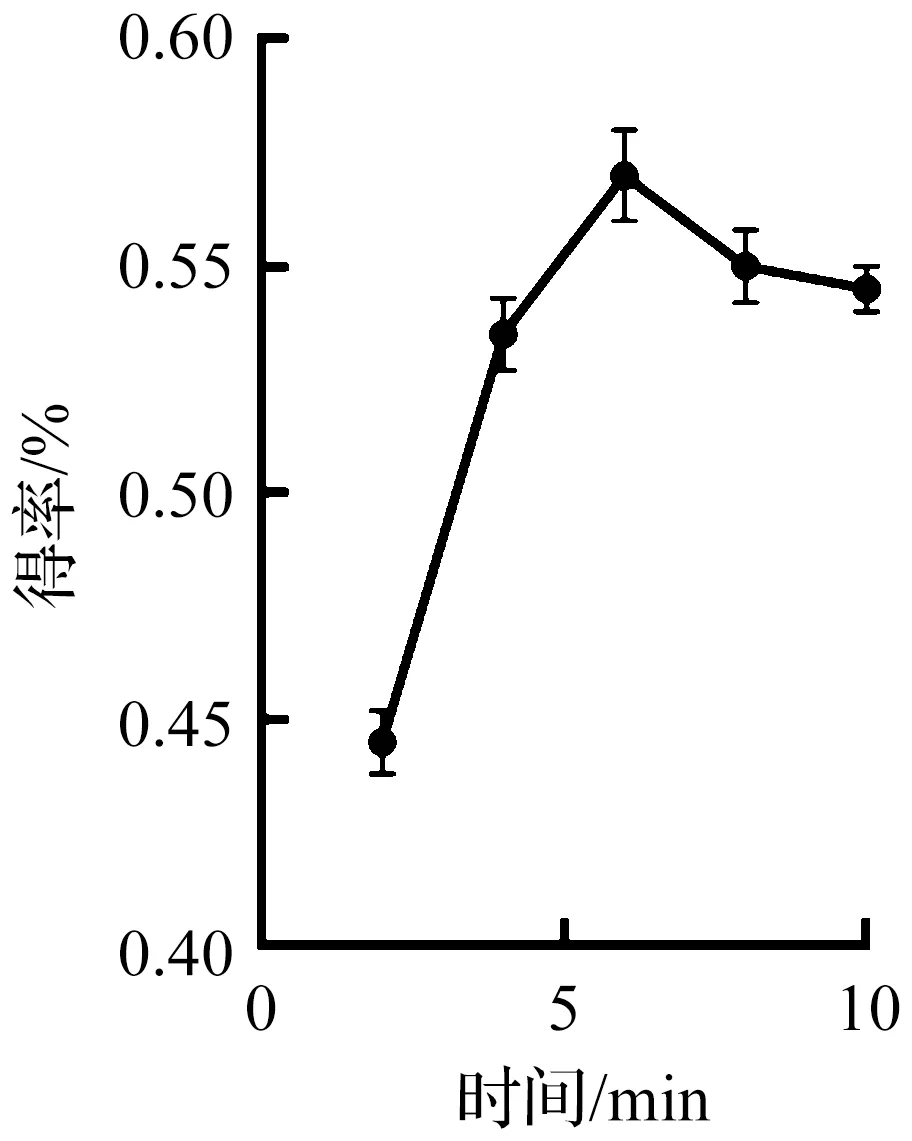
圖3 提取時間對靈芝總三萜得率的影響
2.2.2 料液比對靈芝總三萜得率的影響
當提取時間為6 min,乙醇體積分數為95%時,考察不同料液比1∶20,1∶25,1∶30,1∶35,1∶40 g/mL對靈芝總三萜得率的影響規律,結果如圖4所示。隨著料液比從1∶20 g/mL到1∶35 g/mL,靈芝總三萜的得率出現逐漸上升的趨勢,而且在料液比為1∶35 g/mL時得率呈現最大值,主要是由于提取溶劑用量的增加提高了原料與溶劑間的濃度差,有利于靈芝三萜向溶劑中擴散[19]。當料液比超過1∶35 g/mL時,大部分靈芝三萜已經滲出,靈芝總三萜的得率便不再增加,因此料液比選擇為1∶35 g/mL。
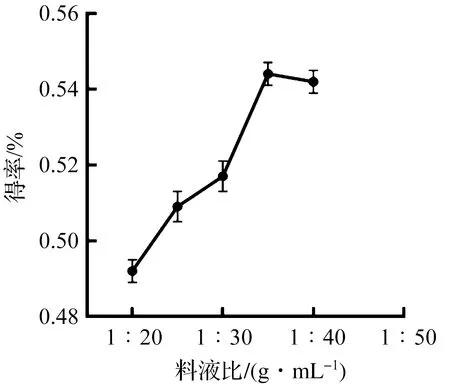
圖4 料液比對靈芝總三萜得率的影響
2.2.3 乙醇體積分數對靈芝總三萜得率的影響
在提取時間為6 min,料液比為1∶35 g/mL的實驗條件下,考察不同乙醇體積分數70%,75%,80%,85%,90%對靈芝總三萜得率的影響規律,結果如圖5所示。隨著乙醇體積分數的提高,靈芝總三萜的得率先上升后趨于穩定,乙醇體積分數從70%增加到85%,提取溶劑體積分數的提高使原料與溶劑充分接觸,有利于靈芝三萜在溶劑中的溶解,因此靈芝總三萜得率逐漸升高,而且在乙醇體積分數為85%時得率呈現最大值,但乙醇體積分數超過85%后,溶劑的極性降低,一些醇溶性成分和極性較小的成分溶出,阻礙目標成分的溶出[19],靈芝總三萜得率趨于穩定,因此乙醇體積分數選擇為85%。
2.3 響應面實驗結果
2.3.1 Box-Behnken實驗設計結果
以靈芝總三萜得率Y為響應值,共計進行17組實驗研究,其中5組中心重復性實驗,響應面實驗方案設計方案及實驗結果如表3所示。響應面設計回歸方程各項殘差分析結果如表4所示。由表4可知:回歸模型顯著性達到顯著性水平,失擬不顯著(P=0.252 9>0.05),說明該模型擬合度良好。復相關系數R2=0.965 4,說明所選因素能夠反映響應值96.54%的變化。回歸模型一次項A,B,C以及二次項A2,C2均有顯著統計學差異,二次項B2有極顯著統計學差異。所得方程:Y=0.56+0.027A+0.033B+0.031C-4.000E-0.003AB+0.014AC-0.018BC-0.039A2-0.074B2-0.043C2
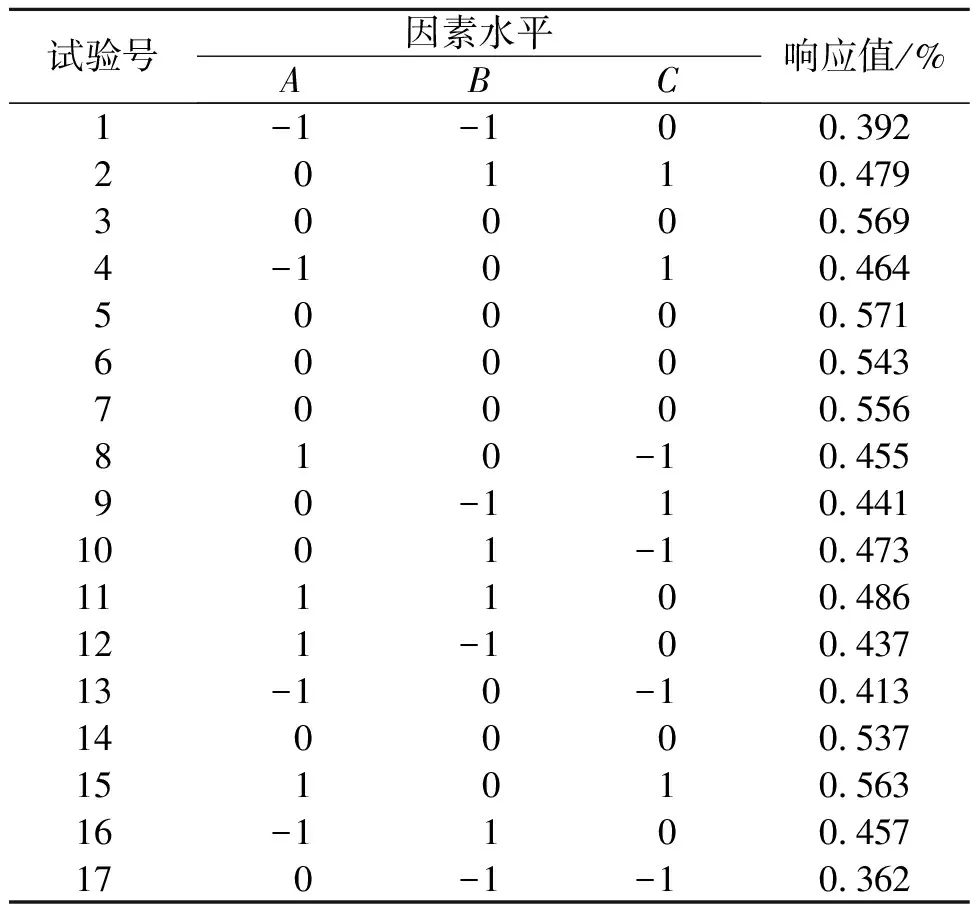
表3 響應面設計方案及實驗結果
2.3.2 響應面分析
根據Design-expert V 10.0回歸分析結果,分別得到低溫剪切提取靈芝總三萜各因素間交互作用的響應面曲線圖,如圖6所示。3D曲面圖上的傾斜率越大,表示兩個因素相互作用越顯著,平面圖上顏色跨度越大,說明響應值變化越快。隨著提取時間A、料液比B及乙醇體積分數C的增加,響應曲面圖上的傾斜率均較大,表明總三萜得率有明顯變化,說明這3個因素的交互作用對總三萜得率的影響較大。響應面上等高線的中心在-1~1時,表示提取靈芝總三萜的最佳工藝條件在設計因子水平范圍內。運用Design-expert V 10.0對回歸模型進行進一步解優化分析,得到靈芝總三萜最高得率的最佳工藝參數:提取時間4.824 min,料液比1∶28.726 g/mL,乙醇體積分數80.420%,靈芝總三萜得率達到0.569%。
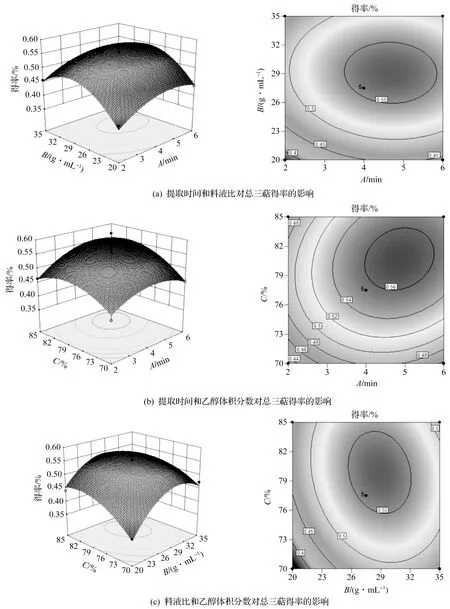
圖6 響應面試驗的相互作用圖
2.3.3 驗證實驗
工藝參數修正為提取時間6 min,料液比1∶30 g/mL,乙醇體積分數80%,做3組平行實驗,所得總三萜得率的平均值為(0.54±0.05)%,與回歸方程所得的總三萜得率0.569%相符,這說明回歸方程能夠真實地反映各因素的交互作用對靈芝總三萜得率的影響,因此用響應面法優化靈芝總三萜得率的回歸模型可靠。
2.4 降血糖活性比較
靈芝酸性三萜,中性三萜以及總三萜的α-葡萄糖苷酶抑制活性如圖7所示。
圖7中:*P<0.05,**P<0.01,***P<0.001,****P<0.000 1。由圖7可知:隨著劑量質量濃度的增加,樣品對α-葡萄糖苷酶活性的抑制能力逐漸增加,因此,低溫高速剪切提取法和傳統乙醇熱回流法提取的靈芝總三萜、中性三萜以及酸性三萜均顯著抑制α-葡萄糖苷酶活性。在相同樣品濃度下,只有CA對α-葡萄糖苷酶活性的抑制能力顯著高于HA(P<0.05),這說明低溫高速剪切技術能夠保護酸性三萜中具有降血糖活性的組分不受破壞,顯著提高酸性三萜的降血糖活性。
靈芝酸性三萜,中性三萜以及總三萜的半抑制濃度(IC50)如表5所示。由表5可知:降血糖能力由大到小排序為中性三萜>總三萜>酸性三萜>阿卡波糖(Acarbose),表明天然產物靈芝三萜的降血糖活性顯著高于現行降血糖藥物阿卡波糖(P<0.000 1)。
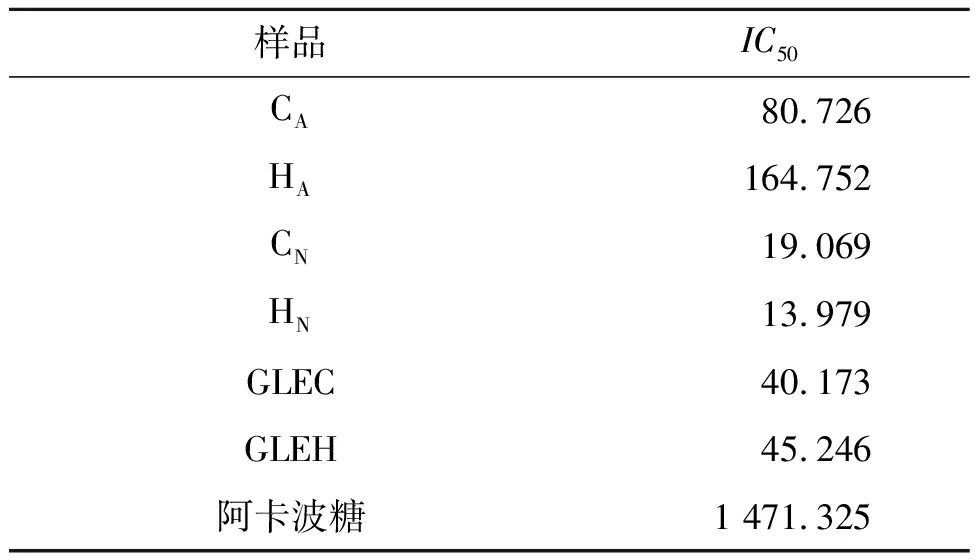
表5 靈芝酸性三萜,中性三萜以及總三萜的半抑制濃度(IC50)
3 結 論
以靈芝子實體為原料,以總三萜得率為評價指標,在單因素實驗的基礎上,通過響應面實驗優化低溫高速剪切法提取靈芝總三萜的最佳工藝參數:提取時間6 min、料液比1∶30 g/mL、乙醇體積分數80%,在此條件下總三萜得率為(0.54±0.05)%,與乙醇熱回流法提取靈芝總三萜得率0.63%接近[11]。將GLEC,CA,CN與GLEH,HA,HN進行α-葡萄糖苷酶活性抑制能力的比較,實驗結果表明:相較于乙醇熱回流法,低溫高速剪切法能顯著提高酸性三萜的α-葡萄糖苷酶活性抑制率。因此,低溫高速剪切技術能快速提取靈芝總三萜,顯著提高酸性三萜的降血糖活性,靈芝總三萜得率接近傳統提取法,為低溫高速剪切技術在食品加工提取領域的應用提供參考。
猜你喜歡
作文·小學低年級(2025年2期)2025-02-13 00:00:00
小雪花·小學生快樂作文(2024年11期)2024-12-31 00:00:00
作文·小學低年級(2024年2期)2024-04-29 00:00:00
作文·小學低年級(2023年3期)2023-04-29 00:00:00
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
小主人報(2022年4期)2022-08-09 08:52:06
中學生數理化·中考版(2022年11期)2022-02-16 07:01:20
小哥白尼(趣味科學)(2019年6期)2019-10-10 01:01:50
發明與創新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
