甲基嘧啶磷原藥中殘留溶劑氣相色譜分析
2023-06-27 12:06:40王勝得曹金艷杜升華
農藥科學與管理 2023年5期
吳 曼,王勝得,曹金艷,陳 明,杜升華
(1.湖南化工研究院有限公司 國家農藥創制工程技術研究中心,湖南 長沙 410014;2.農用化學品湖南省重點實驗室,湖南 長沙 410014)
農藥產品中的雜質情況,是判定農藥產品質量的重要指標。雜質不僅會影響農藥產品質量和使用效果,還可能會對植物、人和動物、環境以及農產品安全產生重要影響[1]。雜質作為農藥的一項關鍵質量屬性,一般包括相關雜質和含量大于0.1%的普通雜質[2]。雜質中的殘留有害溶劑,因沸點低、易揮發、毒性大,需要對其殘留水平進行嚴格控制。本文以甲基嘧啶磷原藥中的常見殘留有機溶劑為例,建立了可與相關雜質同步測定的甲醇、甲基異丁基甲酮(以下簡稱為MIBK)和甲苯的定量檢測方法,旨在對農藥原料藥中殘留溶劑的監測與控制提供借鑒意義。
本文參考農藥中有害溶劑測定常見的氣相色譜法[3,4],考慮同時測定甲基嘧啶磷原藥中其他相關雜質和殘留溶劑的實際工作需求,建立了加相對校正因子的氣相色譜內標法,既可同時測定甲基嘧啶磷原藥中微量殘留溶劑甲醇、MIBK和甲苯含量,也無需每次跟隨溶劑對照品,可實現簡單快速準確的多雜質同步質量控制模式。所述方法對原藥中的殘留有機溶劑和其他相關雜質均做到了有效分離,以相對保留時間進行溶劑定性,以加相對校正因子的內標法進行定量檢測,適用于工廠生產車間的快速品質保障檢測。
1 實驗部分
1.1 儀器和試劑
1.1.1 儀器 島津GC-2010 plus型氣相色譜儀,配有FID檢測器和AOC-20i型自動進樣器。
1.1.2 試劑 甲基嘧啶磷標準品(99.0%);4,4’-二甲氧基二苯甲酮標準品(99.0%);甲醇、MIBK、甲苯均為色譜純(≥99.8%);甲基嘧啶磷原藥(≥95.0%)。
1.2 色譜條件 色譜柱:Agilent DB-17,15 m×0.25 mm(id)熔融石英毛細管柱,膜厚0.5 μm;程序升溫,初始溫度50 ℃,保留2 min,以 20 ℃/min速率升溫至240 ℃并保留5 min,再以30 ℃/min速率升溫至295 ℃并保留10 min;FID檢測器溫度:295 ℃;進樣口溫度:230 ℃;分流比:25∶1;載氣:氦氣,70 kPa,壓力控制模式;進樣量:1 μL.
1.3 溶液的配制
1.3.1 內標溶液的配制 準確稱取內標(4,4’-二甲氧基二苯甲酮)約0.6 g(精確至0.000 2 g)至50 mL容量瓶中,加45 mL乙腈超聲溶解10 min,冷卻后用乙腈定容至刻度,搖勻備用(如有沉淀需過濾)。
1.3.2 混合標準儲備液的配制 準確稱取甲基嘧啶磷標準品、甲醇、MIBK和甲苯標準品各0.06 g(精確至0.000 L2 g)至25 mL容量瓶中,乙腈溶解,定容至刻度,搖勻備用。
1.3.3 混合標準溶液的配制 精密移取標準品儲備液0.5 mL至25 mL容量瓶,加入1 mL上述內標溶液,用乙腈稀釋至刻度,搖勻備用。
1.3.4 試樣溶液的配制 稱取約0.6 g(精確至0.000 2 g)甲基嘧啶磷原藥樣品至25 mL容量瓶中,準確加入上述1 mL內標溶液,用乙腈定容至刻度,搖勻備用。
1.4 樣品測定 在上述儀器操作條件下,待儀器穩定后,連續注入數針混合標準溶液,直至相鄰2針甲基嘧啶磷(或甲醇、MIBK、甲苯)與內標的峰面積比值相對變化不超過1%后,再按照試樣溶液、試樣溶液、混合標準溶液、試樣溶液、試樣溶液、混合標準溶液的進樣順序進行測定。
1.5 計算 殘留溶劑的含量Xi(%)按下式進行計算,其中f和RFi值的計算根據其線性方程的斜率進行計算,詳見2.3項。
式中:Ai為試樣溶液中待測殘留溶劑峰面積,Ais為試樣溶液中內標物峰面積,C為試樣濃度,g/mL,Cis為試樣溶液中內標物濃度,g/mL,f為甲基嘧啶磷校正因子,RFi為殘留溶劑相對于甲基嘧啶磷的相對校正因子。
2 結果與討論
2.1 甲基嘧啶磷原藥中甲醇、MIBK和甲苯的色譜分析圖 甲醇、MIBK和甲苯殘留水平較高的甲基嘧啶磷原藥典型氣相色譜圖(圖1)。由圖可見,甲基嘧啶磷原藥中除殘留有機溶劑外,還含有多種相關雜質。現行殘留溶劑分析方法常見的頂空進樣GC法[5],可避免樣品本身和樣品中的非揮發組分注入氣相色譜儀中造成污染。筆者選用了直接進樣的氣相色譜法,可同時分析甲基嘧啶磷原油中殘留有機溶劑和相關雜質(本文只討論殘留溶劑分析結果);并且在對比了多種不同固定液類型毛細管柱和操作條件分離效果的基礎上,確定了1.2所述色譜操作條件。該法保證殘留溶劑和相關雜質能夠在同一色譜條件下全部流出并有效分離,滿足了運行一針樣品即可完成多組分同步質量控制模式的需求,大大節約了分析時間。
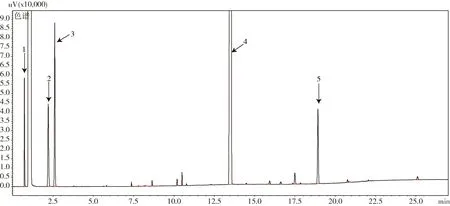
1-甲醇;2-甲基異丁基酮;3-甲苯;4-甲基嘧啶磷;5-內標
2.2 內標物的選擇 本研究對比了正二十二烷、鄰苯二甲酸二乙酯和4,4’-二甲氧基二苯甲酮等一系列內標物,最終選擇4,4’-二甲氧基二苯甲酮(峰5)作為殘留溶劑分析的內標物,雖保留時間距離低沸點溶劑較遠,但是與圖中其他相關雜質完全分離,不與待測組分發生任何化學反應,并且對于同時分析甲基嘧啶磷原油中相關雜質甚至其他高沸點有機溶劑,都是較為理想的內標物。
2.3 線性相關性及校正因子、相對保留時間的測定 線性范圍根據實際測定所需濃度范圍來確定。精確量取標準品儲備液2至10 mL容量瓶中,乙腈稀釋,作為標準品稀釋液。分別從標準品稀釋液中吸取0.1 mL、標準品儲備液中吸取0.1、0.5、2.5、12.5 mL至25 mL容量瓶中,加入1 mL上述內標溶液,用乙腈稀釋至刻度,配制出在2 ~ 1 250 mg/L濃度范圍內系列不同質量濃度的標準溶液,在上述氣相色譜分離條件下進行測定。
以標準品與內標物的質量濃度比為橫坐標,標準品與內標物的峰面積比為縱坐標,分別測定甲基嘧啶磷、甲醇、MIBK和甲苯的線性方程及其相關系數γ,以其線性方程的斜率計算甲醇、MIBK和甲苯相對于甲基嘧啶磷的相對校正因子RFi,RFi=fi/f甲基嘧啶磷=k甲基嘧啶磷/ki,fi=1/ki,f甲基嘧啶磷=1/k甲基嘧啶磷,其中f甲基嘧啶磷和k甲基嘧啶磷為濃度范圍內甲基嘧啶磷的校正因子和線性方程斜率,fi和ki為濃度范圍內甲醇(或MIBK、甲苯)的校正因子和線性方程斜率。以各濃度點色譜峰平均保留時間tR計算甲醇、MIBK和甲苯相對于甲基嘧啶磷的相對保留時間,RRT= tR溶劑/tR甲基嘧啶磷。線性關系(圖2),相對校正因子等(表1)。
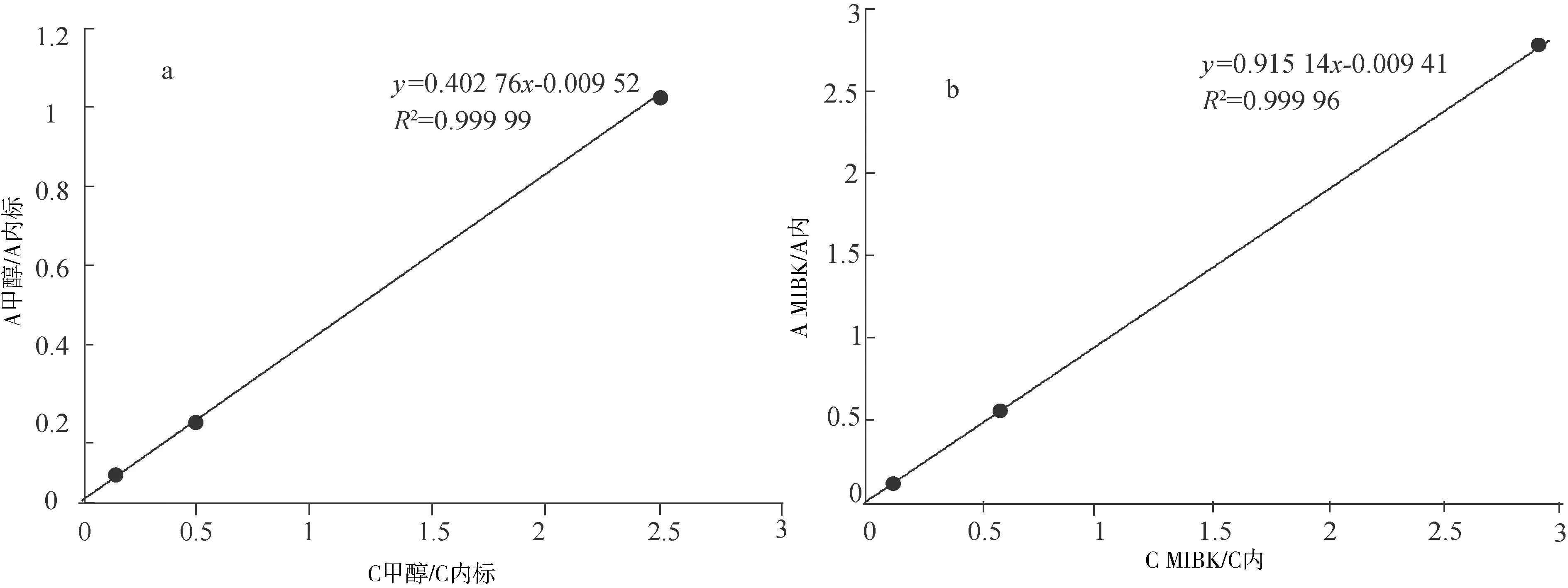
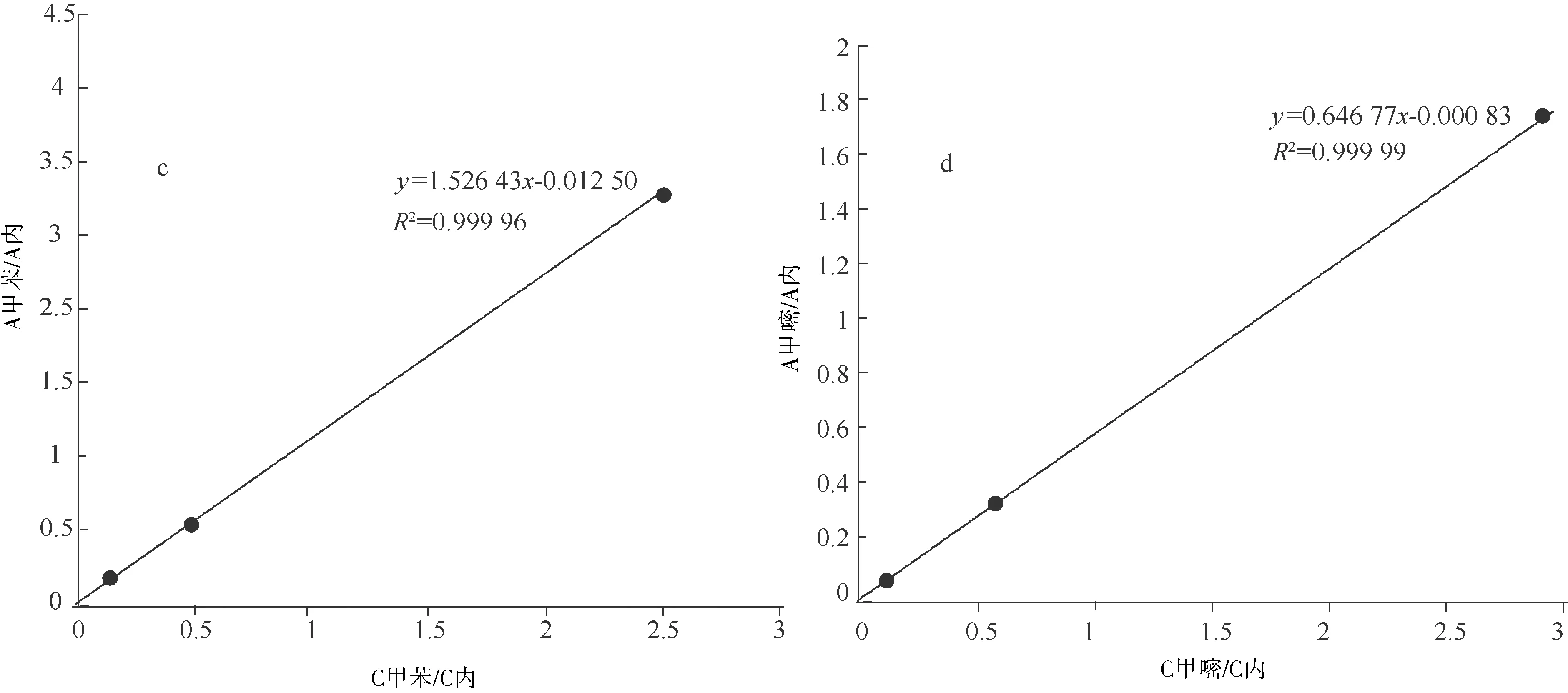
a.甲醇線性關系圖;b.MIBK線性關系圖;c.甲苯線性關系圖;d. 甲基嘧啶磷線性關系圖
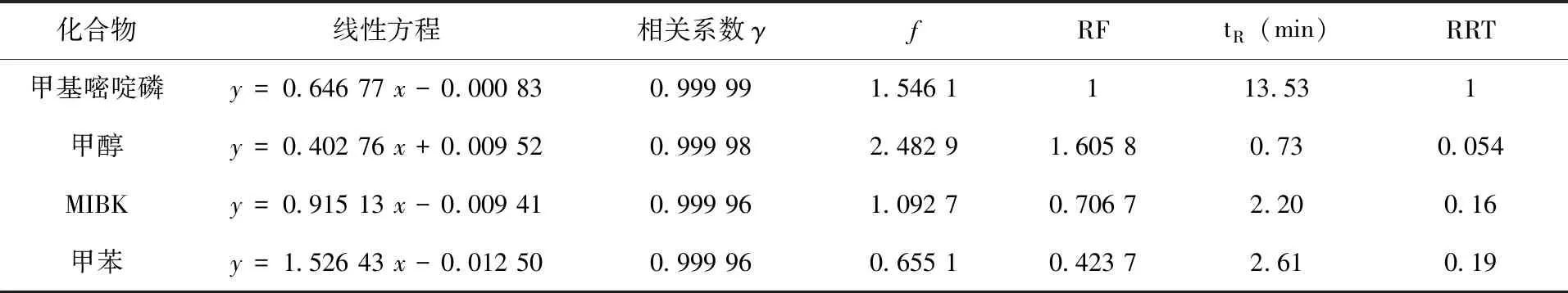
表1 線性關系和相對校正因子、相對保留時間
2.4 分析方法的精密度試驗 在上述氣相色譜操作條件下,對同一樣品中的甲醇、MIBK和甲苯進行6次重復測定,計算其精密度,結果(表2)。甲醇的標準偏差為0.000 89,相對標準偏差為0.99%,MIBK的標準偏差為0.012,相對標準偏差為0.62%,甲苯的標準偏差為0.000 75,相對標準偏差為1.19%,表明儀器操作條件和分析方法精密度高,重現性好,滿足該甲基嘧啶磷原藥中殘留有機溶劑的定量要求。
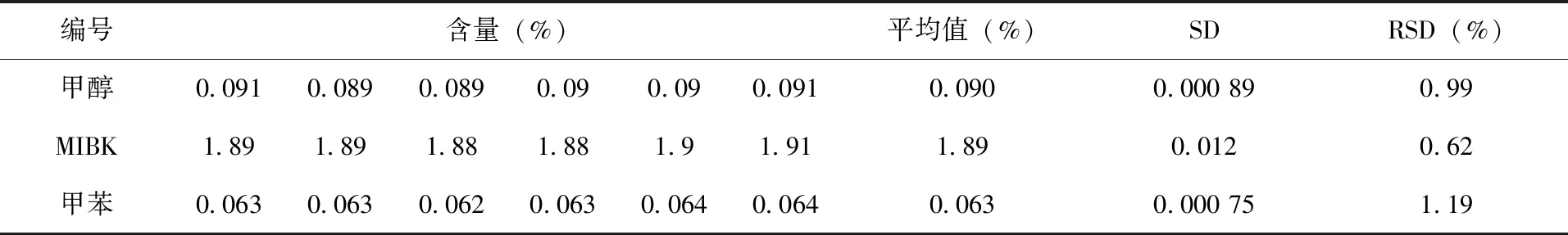
表2 甲醇、MIBK和甲苯精密度測定結果
2.5 分析方法的準確度試驗 稱取適量的甲基嘧啶磷原藥(已測定甲醇、MIBK和甲苯含量),準確加入標準品儲備液,制備成5組樣品,按照上述氣相色譜操作條件測定甲醇、MIBK和甲苯含量,計算回收率,結果(表3)。

表3 回收率測定結果
由表3可知,該方法甲醇、MIBK和甲苯的平均添加回收率為99.07%、100.58%和100.92%,表明儀器操作條件和分析方法可準確測定有效成分含量,滿足雜質定量分析要求[6]。
2.6 外標法比較 用外標法和加相對校正因子的內標法同時測定3批不同批次的甲基嘧啶磷原藥中殘留甲醇、MIBK和甲苯含量。試樣按照1.3.4配制進行測定,用加相對校正因子的內標法計算溶劑含量,另取甲醇、MIBK和甲苯對照品溶液進行測定,用外標法計算試樣中甲醇、MIBK和甲苯含量,結果(表4)。外標法比對結果表明,加相對校正因子的GC內標法能夠準確測定甲基嘧啶磷原藥中殘留溶劑甲醇、MIBK和甲苯含量。
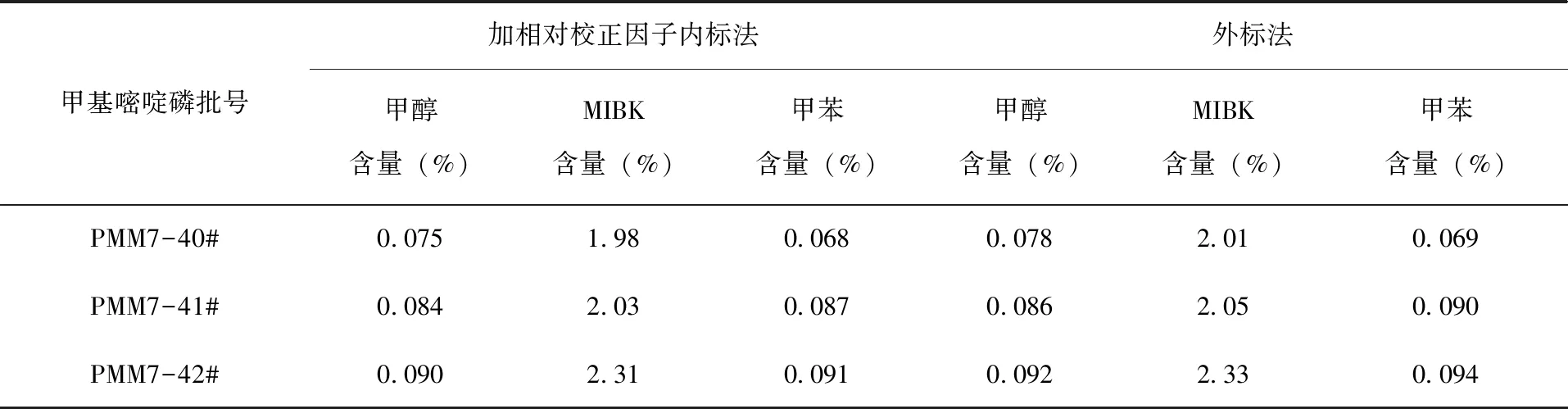
表4 樣品中殘留溶劑含量
3 結論
本文建立的加相對校正因子的氣相色譜內標法,可在同一色譜條件下同時測定甲基嘧啶磷原藥中微量有機溶劑甲醇、甲基異丁基甲酮和甲苯的含量,經線性范圍、精密度和準確度的方法驗證和外標法比較,表明結果準確,方法簡單,可引申為其他殘留有機溶劑和相關雜質的定量檢測方法,僅需測定其對甲基嘧啶磷的相對校正因子,并定期進行相對校正因子的修正即可,可代替外標法,提高實驗效率,對農藥原料藥研究開發的多雜質同步控制提供了參考意義。
