兒童肝豆狀核變性的臨床資料與ATP7B基因分析
2023-07-31 03:26:48彭曉康劉小乖李瑞娜
中國婦幼健康研究 2023年7期
關鍵詞:癥狀
劉 攀,舒 暢,唐 麗,彭曉康,劉小乖,李瑞娜
(西安交通大學附屬兒童醫院感染科,陜西 西安 710003)
肝豆狀核變性(Wilson′s disease,WD)是一種由ATP7B基因缺陷導致的銅代謝障礙性疾病[1-2],是目前少數可治療的遺傳代謝性疾病之一[3]。WD患病率為(0.25~4.00)/10 000,但理論發病率遠高于此[4-5]。ATP7B基因編碼的ATP7B蛋白屬于高度保守的P型三磷酸腺苷(adenosine-triphosphate,ATP)酶超家族的1B類,負責將銅轉運入高爾基體或毛細膽管內,在銅的排泄中起主要作用。ATP7B基因變異導致ATP7B酶活性改變,從而引起肝細胞內銅含量增加,造成肝細胞死亡和銅滲漏入血,進一步引起肝、腦、眼睛、腎臟等組織器官的損傷[2,4]。WD的臨床表現復雜多樣,可為無癥狀性轉氨酶升高、急慢性肝炎、肝硬化、肝衰竭,也可表現為急性溶血性貧血、神經精神癥狀等[1,3-4]。本研究對近年來在西安交通大學附屬兒童醫院就診的158例WD患兒的臨床特征與ATP7B基因進行總結分析,以提高臨床醫師對該病的認識,并進一步探討本地區WD患兒的遺傳學特征。
1資料與方法
1.1研究對象
回顧性收集2016年1月至2022年6月期間收治于西安交通大學附屬兒童醫院感染科的158例診斷明確且為初診的WD患兒作為研究對象。診斷標準:根據中華醫學會肝病學會分會遺傳代謝性肝病協作組制定的《肝豆狀核變性診療指南(2022年版)》[4],推薦應用2001年萊比錫第8屆WD國際會議的診斷標準即Leipzig評分系統[6],總分≥4分可確診。本研究經西安市兒童醫院倫理委員會批準(批準文號:論20220073),并豁免患兒及家屬知情同意。
根據WD患兒的受累器官及臨床表現的不同,分為肝型WD組和腦型WD組,分別有144例、14例。根據肝臟受累的輕重程度及病程長短、表現的差異,肝型WD組又分為癥狀前WD、急慢性肝炎型WD,分別有114例和30例。
1.2資料收集
通過我院電子病歷系統,回顧性收集研究對象初診時的臨床表現、輔助檢查,包括血尿常規、肝功、銅藍蛋白(ceruloplasmin,CER)、24h尿銅、血清銅、凝血功能、腹部超聲及頭顱影像學、基因檢測和眼科角膜K-F環等結果。
1.3 ATP7B基因檢測
經患兒監護人簽署書面知情同意書后,采集患兒及父母靜脈血3mL,提取基因組DNA,并構建與代謝性肝病相關目標基因(ABCB4、ATP7B、CCDC115、NBAS、TMEM199等共573個基因)的全基因組文庫。利用液相捕獲試劑盒捕獲目標基因后進行高通量測序,通過生物信息學分析找出相關基因的變異信息。當疑似有外顯子缺失、臨床高度懷疑WD但未檢測出變異基因或僅檢測到1個變異時,行多重連接依賴探針擴增或全外顯子測序進一步分析。根據獲得的變異結果對患兒及其父母進行Sanger測序驗證。采用生物信息分析軟件PolyPhen-2(http://genetics.bwh.harvard.edu/pph2)、SIFT(http://sift.jcvi.org)及MutationTaster (http://mutationtaster.org)對新變異基因進行致病性預測。依據美國醫學遺傳學及基因組學學會(Ame-rican College of Medical Genetics and Genomics,ACMG)的遺傳變異分類標準與指南[7-8],將變異基因歸類為致病的、可能致病的、意義不明確的、可能良性的或良性的變異。
1.4統計學方法
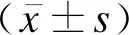
2結果
2.1一般情況
158例WD患兒中男性89名,女性69名,男女比為1.29∶1。WD患兒來自于148個非血緣關系家庭,17例有明確的肝病或WD家族史,其中6例為先證者的家系篩查中發現;確診年齡為6.33(4.03~9.67)歲,其中最小診斷年齡為1.21歲,病程為1.00(0.42,4.75)個月。
癥狀前WD、急慢性肝炎型WD與腦型WD患兒的性別差異無統計學意義(P>0.05),癥狀前WD患兒的確診年齡明顯小于急慢性肝炎型WD與腦型WD(Z值分別為-56.640、-64.956,P<0.05),急慢性肝炎型WD患兒的病程短于癥狀前WD患兒(Z=26.930,P<0.05),見表1。
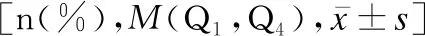
表1 肝型WD組與腦型WD組患兒的一般情況與臨床表現比較
2.2臨床表現
①以肝臟為首發表現的患兒(肝型WD組)有144例,占91.14%,其中無癥狀患兒114例,占72.15%,僅表現為銅代謝指標異常者有4例,轉氨酶升高者110例;查體發現肝臟腫大者36例,其中4例伴脾臟腫大。98例患兒行眼科檢查,其中5例存在角膜K-F環。②以急性慢性肝炎表現者30例,均有轉氨酶升高,其中28例出現不同程度的黃疸(27例為膽汁淤積,1例入院時為高膽紅素血癥、住院第2天復查轉為膽汁淤積),24例存在腹脹或腹痛,21例存在腹腔積液,21例出現肝臟腫大、肝硬化,20例伴有脾臟腫大,21例出現急性肝衰竭,其中18例伴有Coomb′s陰性溶血性貧血。29例患兒行眼科檢查,其中24例存在角膜K-F環。見表1。
以神經精神癥狀為主要表現的WD(腦型WD)患兒有14例,10例有吐字不清、流涎,8例有肢體抖動、震顫,4例有肌張力增高,3例有步態異常,3例有吞咽、咀嚼障礙,3例有情緒低落、表情淡漠,2例有輕度黃疸,2例有轉氨酶輕至中度升高。13例患兒行眼科檢查,均存在角膜K-F環。
癥狀前WD、急慢性肝炎型WD與腦型WD患兒在是否存在K-F環、轉氨酶升高、黃疸、腹脹或腹痛、肝臟腫大、脾臟腫大、腹腔積液、肝硬化、溶血性貧血及神經精神癥狀方面差異均有統計學意義(P<0.05),見表1。
2.3實驗室檢查
158例WD患兒的CER值為0.03(0.01,0.06)g/L,24h尿銅為177.00(115.00,289.60)μg/24h,血清銅為9.87(7.36,13.19)μmol/L。癥狀前WD、急慢性肝炎型WD、腦型WD患兒的總膽紅素(total bilirubin,TB)、直接膽紅素(direct bilirubin,DB)、丙氨酸氨基轉移酶(alanine aminotransferase,ALT)、尿銅、血小板(platelet,PLT)差異均有統計學意義(H值分別為70.150、68.443、60.504、39.502、51.343,P<0.05),見表2。進一步兩兩比較發現:癥狀前WD患兒的血清銅水平較急慢性肝炎型WD患兒低(Z=-27.313,P<0.05),24h尿銅較急慢性肝炎型WD、腦型WD患兒低(Z值分別為-54.929、-33.005,P<0.05)。腦型WD患兒的天冬氨酸氨基轉移酶(aspartate aminotransferase,AST)、白細胞(white blood cell,WBC)較癥狀前WD與急慢性肝炎型WD患兒低(AST:Z值分別為51.234、52.013;WBC:Z值分別為39.033、38.981,P<0.05)。急慢性肝炎型WD患兒的血清白蛋白(albumin,Alb)、堿性磷酸酶(alkaline phosphatase,ALP)、紅細胞計數(red blood cells,RBC)、血紅蛋白(hemoglobin,Hb)較癥狀前WD、腦型WD患兒低(Alb:Z值分別為69.710、-40.250;ALP:Z值分別為54.067、-34.673;RBC:Z值分別為64.734、-52.923;Hb:Z值分別為62.763、-64.000,P<0.05);CER水平、γ-谷氨酰轉肽酶(γ-glutamyl transpeptidase,GGT)、較癥狀前WD、腦型WD患兒高(CER:Z值分別為-32.257、36.000;GGT:Z值分別為-32.571、54.154,P<0.05)。
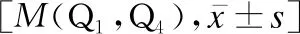
表2 肝型WD與腦型WD組患兒的實驗室檢查比較
2.4 ATP7B基因
158例WD患兒中有116例進行了基因檢測,基因檢測率為73.42%,癥狀前WD患兒90例,急慢性肝炎型WD患兒21例,腦型WD患兒5例;其中5例為雜合變異,14例為純合變異,97例為復合雜合變異(8例有3種變異類型)。共檢測出64種變異,包括錯義突變39種、同義變異3種、無義變異4種、移碼變異8種、剪切變異5種、缺失變異5種(1種單個氨基酸缺失,4種外顯子缺失)。熱點突變前4位依次為c.2333G>T(p.R778L)、c.2621C>T(p.A874V)、c.2310C>G(L770=)及c.2975C>T(p.P992L),等位基因頻率分別為28.88%、9.48%、8.62%、7.76%。發現6種新的未報道的基因變異,包括c.1908dupC(p.Asn637Glnfs*118)、c.4179_4180insC(p.Pro1394 Profs*15)、c.1604A>G(p.Glu535Gly)、c.2278C>T(p.Pro760Ser)、c.3008C>A(p.Ala1003Glu)及c.3532A>C(p.Thr1178Pro)。根據ACMG指南判定,除c.1604A>G(p.Glu535Gly)為意義不明的變異外,其余均為可能致病性變異。
依據患兒是否存在c.2333G>T(p.R778L)變異進行基因型的分組,分為R778L型WD與其他型WD,分別有55例、61例。R778L型WD的確診年齡大于其他型WD(Z=-2.082,P<0.05),而臨床分型、病程、ALT、AST、WBC、Hb、PLT、CER、尿銅、血清銅、K-F環陽性率兩組比較差異均無統計學意義(P>0.05),見表3。

表3 R778L型WD與其他型WD患兒的臨床比較 [n(%),M(Q1,Q4)]
3討論
3.1兒童WD的一般情況與臨床表現
WD是一種銅蓄積性的遺傳代謝病,常以肝臟受累起病,發病年齡相對較早[3]。我院收治的WD患兒中肝型WD占91.14%,其中以僅表現為肝酶升高,伴或不伴有肝腫大的癥狀前WD居多,占72.15%。癥狀前WD患兒多在入園、入學體檢時發現,確診年齡中位數為5.30歲,明顯小于急慢性肝炎型WD與腦型WD的確診年齡(P<0.05)。WD最小診斷年齡為1.21歲,為先證者的家系篩查中發現。WD患兒基因檢測率為73.42%,有部分患兒監護人認為臨床已明確診斷為WD而不需進行基因檢測,這是錯誤的觀點,仍推薦對先證者的直系親屬或有WD家族史者進行家系的基因檢測[4,9-11]。
急慢性肝炎型WD組中有21例急性起病,出現肝功能衰竭,其中18例伴有Coomb′s陰性溶血性貧血。雖然患兒黃疸、腹痛或腹脹等病程短(中位數為0.37個月),但查體多有慢性肝病的異常體征,如肝硬化、腹水等。WD患兒,當肝細胞內的銅超載到一定水平會造成肝臟和其他組織器官損害,但肝臟為沉默器官,無明顯臨床癥狀時多不會引起患兒不適或家長關注,往往就診時已出現代償期甚至失代償期肝硬化。這與Ferenci等人[12]報道的39.50%的兒童及青少年在診斷WD時已存在肝硬化的結果一致。2例患兒出現表情淡漠、震顫的神經精神癥狀,結合血氨異常升高,頭顱磁共振檢查正常,診斷為高血氨癥、肝性腦病。對于肝型WD伴隨有神經精神癥狀的患兒,臨床醫師需注意鑒別肝性腦病與WD神經系統損害表現,以免貽誤病情。
3.2兒童WD的輔助檢查
急慢性肝炎型WD的CER、GGT顯著高于癥狀前WD及腦型WD組,而ALP、ALB、RBC、Hb均較另外兩組明顯下降。急慢性肝炎時肝細胞損傷、細胞膜破壞,細胞內蓄積過量的銅離子、銅藍蛋白前體被釋放入血,銅離子的金屬毒性對紅細胞破壞可造成貧血,腎臟排泄銅的代償作用加強,尿銅升高明顯;肝損害造成肝臟合成功能下降,從而導致白蛋白降低。以急性肝衰竭起病的WD患者,銅藍蛋白可能下降不明顯或正常,早期診斷存在一定困難,臨床醫師仍需提高對WD診斷的警惕性[3,13]。有學者提出,在急性肝衰竭人群中識別WD的順序指標:ALP/TB<4,AST/ALT>2.2,敏感度為94.00%,特異度為86.00%~94.00%[14],但其診斷效能仍有爭議。
3.3兒童WD臨床表型與基因表型的相關性分析
WD患者的ATP7B基因在不同地域、種族和人群的變異特征存在明顯差異,歐洲以p.H1069G為主[15],而亞洲人群以p.R778L最為常見,等位基因頻率為12.00%~39.00%[16-19]。我國WD患者主要以p.R778L、p.P992L和p.T935M變異為主[18],但不同地區的中國人也有不同的ATP7B變異類型,具有區域特異性分布的特征[20]。本研究WD患兒最常見的突變位點為c.2333G>T(p.R778L),等位基因頻率為28.88%;其次為c.2621C>T(p.A874V),等位基因頻率為9.48%,該發現與其他研究報道的中國WD人群的常見突變基因結果相一致[21-22]。
WD患兒的臨床表現多種多樣,臨床表型差異較大,國內外眾多研究均嘗試探索WD的臨床表型與基因型的相關性,但目前尚無統一且公認的結論出現。本研究發現,p.R778L型WD的確診年齡高于其他型WD,而在臨床分型、病程、CER、血清銅等方面兩組差異無統計學意義。該結論與黃帆等[23]報道的“p.R778L突變WD患者的發病年齡較晚,與首發癥狀和銅代謝障礙無關”相符,而與Cheng等[18]報道的“中國WD患者中p.R778L組發病年齡更早,CER水平及血清銅更低”的結論不一致。WD臨床表型與基因型是否相關存在一定爭議,可能與不同地域、樣本量的選取及修飾基因的影響作用等因素有關,仍待大量臨床數據積累,以及多中心、大樣本研究的驗證。
利益沖突聲明:所有作者均聲明不存在利益沖突。
猜你喜歡
初中生學習指導·提升版(2023年8期)2023-09-12 10:26:19
保健醫苑(2022年1期)2022-08-30 08:39:40
中老年保健(2021年12期)2021-08-24 03:30:44
今日農業(2020年17期)2020-10-27 03:10:52
今日農業(2020年16期)2020-09-25 03:05:08
家庭醫學(下半月)(2020年2期)2020-05-11 02:07:18
基層中醫藥(2020年10期)2020-02-13 15:45:52
吉林蔬菜(2017年10期)2017-11-01 07:47:04
獸醫導刊(2016年6期)2016-05-17 03:50:35
中國醫學影像學雜志(2015年9期)2015-12-15 11:03:26
