TpBD(NH2)2的D-葡萄糖衍生物高效液相色譜固定相研究*
2023-08-01 07:59:26李斗發李亞楠劉華林
云南化工 2023年7期
李斗發,李亞楠,劉華林,字 敏
(云南師范大學 化學化工學院,云南 昆明 650500)
引言
在臨床上使用的很多藥品是手性藥物[1]。不同的異構體具有不同的生理活性、物理和化學性質,有些藥品一個對映體有藥效,另一個對映體可能沒有藥效,甚至對人的身體健康有害[2]。所以,對具有手性的藥物進行分離具有重大的現實意義。由于外消旋體的理化性質差別很小,很難把對映異構體分離開[3],分離手性藥物的方法很多,其中比較典型的有毛細管電色譜(SEC)、高效液相色譜(HPLC)、氣相色譜(GC)等[4-6]。手性固定相CSP(Chiral stationary phase,CSP)分離手性化合物是當前比較熱門的研究方向,使用比較廣的是多糖、環糊精和冠醚作為固定相[7-9]。
在過去的十多年里,研究員對共價有機骨架材料COFs(covalent organic frameworks,COFs)做了很多探索和研究,因為其具有很多優良的性能,例如,具有高的表面積、孔的體積、形狀容易調節,方便組裝[10]。COFs大量應用于氣相色譜、液相色譜[11]、催化和識別等[12]領域。
本文使用后合成修飾法[13]和溶劑熱合成法[14],獲得了具有手性的TpBD(NH2)2的D-葡萄糖衍生物,將其采用網包法[15-16]覆蓋在球形硅膠上,用于制備手性固定相。實驗結果表明TpBD(NH2)2的D-葡萄糖衍生物能夠很好地分離手性藥物、氨基酸以及位置異構體。與此同時,制備的手性固定相填充的分離柱具有較好的穩定性。通過研究表明,制備的手性CSP在分離手性化合物具有較大的潛能。
1 實驗部分
1.1 試劑和儀器
D-Max-3B 2000X粉末衍射儀(日本,Rigaku公司);S-3000N掃描電子顯微鏡(日本,Hitachi公司);Chirascan圓二色光譜儀(英國,應用光學物理);BRUKERTensor-27傅里葉紅外變換紅外光譜儀(德國,Bruker);ASAP2020 氮氣吸附儀(美國,Micromeritics公司);EliteP230Ⅱ高壓流壓泵(大連依力特分析儀器有限公司);EliteUV230Ⅱ 紫外檢測儀(大連依力特分析儀器有限公司);AT-330柱溫箱(大連依力特分析儀器有限公司);1666液相色譜裝柱機(美國,Alltechi有限公司);200 mm×φ4.6 mm 不銹鋼高效液相色譜空柱(美國,Alltechi有限公司);UniSil 5-100硅膠(蘇州納微科技股份公司)。
六亞甲基四胺(97%)、三氟乙酸(97%)、鄰硝基苯胺(99%)、碘化鉀(98%)、高碘酸鉀(99%)、位置異構體(99%)均購于上海阿拉丁生化科技股份有限公司;三乙胺(≥99%)、二茂鐵二氯化鈀(99%)、均三甲苯(99%)、無水SnCl2(99%)均購于上海阿達瑪斯試劑公司;氯化鈉(≥99%)、葡萄糖(99%)購于天津市風船化學試劑科技有限公司;1,4二氧六環(98%),外消旋化合物(99%)均購于美國Sigma-Aldrich有限公司;間苯三酚(98%)購于鄭州阿爾法化工有限公司。
1.2 單體1,3,5-三甲醛間苯三酚(Tp)的合成
取用經干燥后的 7.549 g 六亞甲基四胺(54 mmol),3.007 g 間苯三酚(24.5 mmol),加入 500 mL 的三頸燒瓶中。對裝置抽真空后充滿N2,在N2的保護下,邊攪拌邊加入 90 mL 三氟乙酸,后將裝置移入油浴鍋,在 100 ℃ 下攪拌 3 h 后,取用配制好的 0.3 mol/L 的鹽酸溶液 150 mL 加入到反應容器中,再反應 2.5 h。燒瓶取出冷卻后,用二氯甲烷萃取產物,取有機相層,蒸發掉溶液后,用甲醇抽濾洗滌產物,得到單體1,3,5-三甲醛間苯三酚(Tp),產物呈粉色。
1.3 單體3,3′-二硝基聯苯胺(DNB)的合成
取 1.24 g 4-碘-2-硝基苯胺(5 mmol),三乙胺 1.4 mL,0.016 g 二茂鐵二氯化鈀(DPPF,0.5 mmol),17.5 mL 無水甲苯,依次加入到 100 mL 二頸燒瓶中,在90~130 ℃ 下攪拌反應 12 h,得到單體3,3′-二硝基聯苯胺(DNB),產物呈紅色、粉末狀。
1.4 TpBD(NH2)2的合成
稱取 21.0 mg 的Tp(0.1 mmol),41.1 mg 的DNB(0.15 mmol),均三苯甲醛 0.75 mL,1,4-二氧六環 0.75 mL,3 mol/L 醋酸 0.5 mL,于Pyrex耐熱玻璃管中,超聲至其混合均勻。用液氮冷凍后,抽真空脫氣,用酒精噴燈封管,在 120 ℃ 的條件下反應 72 h,待反應完成后,將玻璃管敲碎,取出產物,依次用丙酮、二氯甲烷洗滌,得到呈紅棕色的產物。
取 3 g SnCl2·2H2O和 150 mg 上述合成的TpBD(NO2)2于 50 mL 的二頸燒瓶中,加入 5 mL 無水四氫呋喃使其混合溶解,在 50 ℃ 條件下加熱回流 3 h。反應完成后,將沉淀物用 70 mL 的 1 mol/L 鹽酸洗滌10次,用 70 mL 的水洗滌3次,并用 100 mL 丙酮洗滌一次。然后取上步產物于合適的反應釜中,加入 5 mL 苯甲醚,在 120 ℃ 烘箱內加熱 24 h。最后,將粉末過濾并用 100 mL 丙酮洗滌。得到紅棕色產物 TpBD(NH2)2,合成路線如圖1。

圖1 TpBD(NH2)2的合成路線
1.5 TpBD(NH2)2的D-葡萄糖衍生物的合成
稱取 150 mg 上述合成的TpBD(NH2)2,1 g 干燥后的D-葡萄糖,20 mL 無水甲醇于 50 mL 的二頸燒瓶中,在 70 ℃ 條件下攪拌 4 d,待反應完成后,用純水洗去未反應完的葡萄糖,烘干,得到紅棕色的產物。合成路線如圖2。

圖2 TpBD(NH2)2的D-葡萄糖衍生物的合成路線
1.6 利用網包法制備TpBD(NH2)2的D-葡萄糖衍生物手性固定相
將TpBD(NH2)2的D-葡萄糖衍生物用瑪瑙研缽研磨至比硅膠還細,稱取 1.0 g 哌嗪于 50 mL 容量瓶內,加入異丙醇和 2 mL 三乙胺配制成 50 mL 溶液,稱取 100 mg 研磨好的TpBD(NH2)2的D-葡萄糖衍生物加入到一定量配制好的上述溶液中,超聲后浸泡上述硅膠3~ 4 h,吸出多余液體,在 110 ℃ 烘箱里加熱處理 15 min,備用。
1.7 TpBD(NH2)2的D-葡萄糖衍生物手性固定相色譜柱的填充
稱取 4.2 g 制備好的固定相,用有機膜濾過的正己烷/異丙醇(體積比9∶1)為溶劑,用250目的篩子,將固定相用溶劑濕法過篩,待固定相沉降后,倒出上清液。采用高壓勻漿法裝柱,在裝柱機的溶劑罐中加入約 800 mL 的上述溶劑,向過篩后的固定相加入約 40 m L的上述溶劑,倒入勻漿罐前,為保證固定相分散均勻,要攪拌后立即倒入。擰緊裝置后,保持 35 MPa 5 min,后將壓力調為 25 MPa 保持 30 min,結束后打開減壓閥,取下色譜柱,裝上柱頭,貼上標簽標識上下端,裝到液相色譜儀器上,用正己烷/異丙醇(體積比9∶1)為流動相,0.5 mL/min 的流速沖洗色譜柱,待基線穩定即可測樣。
1.8 色譜拆分條件和性能評價參數
用正己烷/異丙醇(體積比9∶1;8∶2)和甲醇/水(體積比9∶1;8∶2)作為流動相,流速為 0.5 mL/min,紫外檢測波長分別為 254、210,220 nm,柱溫為 25 ℃。
對色譜柱的分離效果用保留因子k,分離因子α和分離度Rs進行評價。公式分別為:
k1=(t1-t0)/t0
(1)
k2=(t2-t0)/t0
(2)
(3)
(4)

2 結果與討論
2.1 1,3,5-三甲醛間苯三酚(Tp)和3,3′-二硝基聯苯胺(DNB)的表征
對1,3,5-三甲醛間苯三酚(Tp)進行核磁共振氫譜表征。1H NMR(500 MHz,CDCl3)δ:10.18(s,3H,CHO),14.15(s,3H,OH)。對DNB進行核磁共振表征。1H NMR(500MHz,DMSO-d6)δ:7.1(d,J=8.9Hz,2H),7.54(s,4H),7.74(dd,J=2.3Hz,2H),8.14(d,2H)。上述所得數據與文獻報道[16]一致,表明已經成功合成Tp和DNB。
2.2 TpBD(NH2)2的D-葡萄糖衍生物手性硅膠球的表征
2.2.1 TpBD(NH2)2的D-葡萄糖衍生物的粉末X衍射表征分析
據文獻報道,TpBD(NO2)2的PXRD圖案在對應于(100)面的3.5°處顯示出強烈衍射,6.0°、9.4°和26°的反射分別歸因于(110)、(210)和(001)平面,屬于AA堆疊模式。由圖3看出,在3.5°、6.0°、9.4°、26°均出現了衍射峰,與文獻報道一致[1-3],表明TpBD(NO2)2成功合成。經還原的TpBD(NO2)2和用D-葡萄糖修飾后的TpBD(NH2)2,其主要衍射峰也在3.5°、6.0°、9.4°、26°處,三者的峰形和出峰的位置基本相似,這也說明還原以及修飾后的物質的晶型和結構特征基本保持不變。

a.TpBD(NO2)2,b.TpBD(NH2)2,c.TpBD(NH2)2的D-葡萄糖衍生物
2.2.2 TpBD(NH2)2的D-葡萄糖衍生物的紅外表征
TpBD(NO2)2、TpBD(NH2)2和TpBD(NH2)2的D-葡萄糖衍生物的紅外表征如圖4所示,從圖4(a)TpBD(NO2)2的紅外圖可以看出,TpBD(NO2)2在 1512 cm-1和 1362 cm-1有吸收峰,對應于-NO2的特征吸收峰。從圖4(b)TpBD(NH2)2的紅外圖顯示,對比圖4(a),在 1512 cm-1和 1362 cm-1處的峰強度減弱,說明TpBD(NO2)2中的-NO2被還原了。觀察圖4(c)在 1069 cm-1左右為C-O的特征吸收峰,對比圖4(b),強度有所增強,說明D-葡萄糖成功修飾了TpBD(NH2)2。

a.TpBD(NO2)2,b.TpBD(NH2)2,c.TpBD(NH2)2的D-葡萄糖衍生物
2.2.3 TpBD(NH2)2的D-葡萄糖衍生物氮氣吸附測試
為了進一步了解合成的D-葡萄糖衍生物的結構特征,我們對其進行了氮氣吸附測試,結果見圖5。使用Brunauer-Emmett-Teller(BET)方法對測量數據進行計算,得到TpBD(NH2)2的D-葡萄糖衍生物的比表面積為 264 m2·g-1,孔體積為 1.78 cm3·g-1,通過Barrett-Joyner-Halenda(BHJ)分析計算測試數據,得到其平均孔徑是 1.92 nm(圖5b),符合相關文獻報道的參數。

圖5 TpBD(NH2)2的D-葡萄糖衍生物的氮氣吸附測試
2.2.4 TpBD(NH2)2的D-葡萄糖衍生物的圓二色譜分析
如圖6所示通過D-葡萄糖修飾后的 TpBD(NH2)2顯示出了Cotton效應,表明葡萄糖成功修飾在COF上,使衍生后的TpBD(NH2)2具有手性。
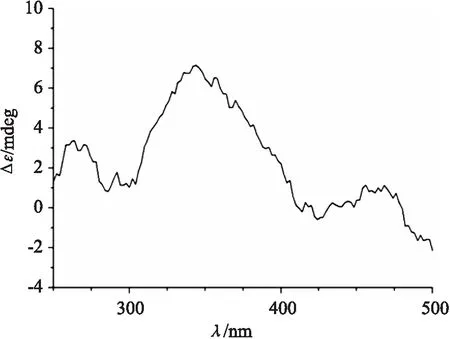
圖6 TpBD(NH2)2的D-葡萄糖衍生物圓二色譜圖
2.2.5 TpBD(NH2)2的D-葡萄糖衍生物的掃描電鏡
為了觀察采用網包法后,TpBD(NH2)2的D-葡萄糖衍生物有沒有固載到硅膠上,分別通過掃描電鏡對空硅膠和網包后的固定相進行觀察,結果見圖7。通過電鏡圖片可以觀察到,沒有經過網包的硅膠表面光滑,沒有任何物質固載在其表面,而經過網包的硅膠,其表面固載了大量的經過研磨的TpBD(NH2)2的D-葡萄糖衍生物,說明通過網包法,所合成的手性COF材料已成功固載在硅膠表面。

(a)純硅膠,(b)TpBD(NH2)2的D-葡萄糖衍生物固定相
2.3 TpBD(NH2)2的D-葡萄糖衍生物手性固定相對于外消旋體的拆分
為了檢測TpBD(NH2)2的D-葡萄糖衍生物制備的色譜柱的拆分能力,用其對外消旋體進行拆分。實驗結果顯示,所制備的手性色譜柱對15種手性物質能夠進行不同程度的分離。其中CH3OH/H2O(體積比9∶1)的流動相對4種手性物質有一定的拆分效果,分別為二苯基乙醇酮、華法林、N-(3,5-二硝基苯甲酰)-L-亮氨酸、1-苯乙醇,而CH3(CH2)4CH3/(CH3)2CHOH的流動相對11種手性物質有一定的拆分效果,它們分別是2-甲基戊酸甲酯、2-乙基己酸、1,2-環氧丁烷、環氧溴丙烷、3-氯-2-丁酮、2-甲基戊醛、3-羥基丁酸乙酯、異丙基縮水甘油醚、乳酸乙酯、1-(1-萘基)乙胺、γ-辛內酯。它們的拆分結果見表1。

表1 色譜柱對15種手性物質的拆分結果
表1的分離結果顯示,在CH3OH/H2O(體積比9∶1)的流動相條件下,所拆分的外消旋體中,二苯基乙醇酮和華法林做到了基線分離,拆分情況比較理想。在CH3(CH2)4CH3/(CH3)2CHOH的流動相條件下,2-乙基己酸、環氧溴丙烷、乳酸乙酯等達到了基線分離。兩種流動相條件下,所拆分的物質不相同,能夠相互補充,拓展了TpBD(NH2)2的D-葡萄糖衍生物手性固定相的拆分范圍,該手性色譜柱所拆分的部分手性物質的譜圖如圖8所示。

圖8 TpBD(NH2)2的D-葡萄糖衍生物色譜柱對外消旋體的部分圖譜
測試結果顯示TpBD(NH2)2的D-葡萄糖衍生物對一些手性物質具有比較好的分離效果,有些甚至能達到基線分離。能夠對手性物質進行拆分的原因是,COFs材料上被修飾了D-葡萄糖手性基團,使COFs材料具備了手性,手性COFs材料對兩種對映異構體存在保留差異,從而能夠對手性化合物進行拆分。除此之外,手性固定相和手性物質之間的各種作用力,可能也在某種程度上對這些物質的分離作出貢獻。
2.4 TpBD(NH2)2的D-葡萄糖衍生物液相色譜柱對于苯系位置異構體的拆分
為了探究TpBD(NH2)2的D-葡萄糖衍生物對于苯系位置異構體的分離情況,我們用所制備的色譜柱對苯系位置異構體進行了實驗。在所分離開的這些物質中,該色譜柱對o,m,p-鄰溴苯胺、o,m,p-硝基苯酚、o,m,p-氯苯酚能夠進行一定程度的分離。它們的拆分結果見表2,拆分譜圖見圖9。

表2 色譜柱對2種位置異構體的拆分結果

圖9 TpBD(NH2)2的D-葡萄糖衍生物液相色譜柱對苯系位置異構體的部分色譜拆分圖
從圖9中可以觀察到,因苯系位置異構體的分子結構存在一些差異,因此導致它們在經過色譜柱時,該固定相對它們的作用不同,使得它們隨著流動相從色譜柱中流出的時間不相同,即彼此的保留時間有差別,這也正是它們能夠被拆分的原因。從分離圖譜可知,TpBD(NH2)2的D-葡萄糖衍生物色譜柱對3種苯系位置異構體都有一定的拆分效果,這也拓展了TpBD(NH2)2的D-葡萄糖衍生物手性固定相在物質分離方面的應用。
2.5 溫度對TpBD(NH2)2的D-葡萄糖衍生物色譜柱的影響
為了研究不同溫度對TpBD(NH2)2的D-葡萄糖衍生物色譜柱出峰情況的影響,選用華法林進行測試。探究了25、30、35、40、45 ℃ 五個檢測溫度對TpBD(NH2)2的D-葡萄糖衍生物手性固定相出峰效果的影響。從對比譜圖(圖10)中可見,隨著溫度的升高,其保留時間在逐漸往后移動。因此可知,在一定溫度范圍內,隨著溫度的升高,對色譜柱的出峰情況沒有太大影響,只有保留時間在后移,不影響出峰情況。

圖10 不同溫度下華法林在TpBD(NH2)2的D-葡萄糖衍生物在色譜柱上的分離譜圖.
2.6 TpBD(NH2)2的D-葡萄糖衍生物色譜柱的重復性
為了探討TpBD(NH2)2的D-葡萄糖衍生物所制備的色譜柱的重復性,選擇華法林為代表,重復測樣5次,其拆分譜圖如圖11所示,經過計算,第一個峰的峰面積與保留時間的相對標準偏差(RSD)分別為1.34%和1.25%。這一結果表明,TpBD(NH2)2的D-葡萄糖衍生物所制備的色譜柱具有良好的重復性。

圖11 華法林在TpBD(NH2)2的D-葡萄糖衍生物色譜柱上的重復性分離譜圖
3 結論
本文通過查閱相關文獻制備了COFs結構 TpBD(NH2)2,使用后合成修飾的方法,讓葡萄糖修飾到了該材料上,通過“網包法”固載到球型硅膠表面,獲得了高效液相色譜手性固定相。用該固定相對二苯基乙醇酮、華法林和N-(3,5-二硝基苯甲酰)-L-亮氨酸等15種手性物質進行了分離。研究結果表明,本文合成的手性材料能夠使多種手性物質實現不同程度的分離。其中CH3OH/H2O(體積比9∶1)的流動相對4種手性物質起到一定的拆分作用,CH3(CH2)4CH3/(CH3)2CHOH(體積比9∶1)的流動相對11種手性物質有一定的拆分效果。這些結果證明,采用后合成修飾的策略,將手性基團連接到無手性的共價有機框架上是可行的,手性的COFs材料用于液相色譜固定相具有一定的前景。
