基于電導率-含水量曲線法制備、優化廣藿香揮發油微乳的研究*
2023-09-13 07:14:08鐘文嘉黃益穗劉灼波
中醫藥導報 2023年8期
鐘文嘉,黃益穗,劉灼波
(廣州中醫藥大學第一附屬醫院,廣東 廣州 510405)
廣藿香揮發油是廣藿香的主要藥用有效成分[1],具有芳香化濁、解暑的功效[2]。現代藥理學表明,廣藿香揮發油具有抗菌、抗炎、調整胃腸運動功能等作用[3]。由于廣藿香揮發油具脂溶性,口服入藥難于在胃腸道被吸收,制成微乳制劑(O/W型)可改善其在胃腸道的吸收性,從而提高生物利用度[4]。微乳制劑是由一定比例的油相、水相、表面活性劑和助表面活性劑配制而成的熱力學穩定體系,其粒徑一般介于10~100 nm之間。要獲得穩定性好的O/W型微乳,找準O/W型微乳成型臨界點尤為重要。目前的報道[5-6]多為目測法,即肉眼觀察體系由混濁至澄清作為O/W型微乳成型臨界點;也有報道[7-9]提到建立電導率-含水量曲線法,以電導率最大值時對應的含水量值作為O/W型微乳成型臨界點,但是沒有采用更為直觀的指標來證實電導率-含水量曲線法的精確性。本研究基于此背景,分別采用目測法、電導率-含水量曲線法確定微乳的O/W型微乳成型臨界點,按各自獲得的臨界點分別配制微乳,通過比較平均粒徑、多分散系數(polydiseperse Index,PDI),證實電導率-含水量曲線法比目測法確定的O/W型微乳成型臨界點更為精確,并在此基礎上,使用該方法制備并優化廣藿香揮發油微乳,以期為中藥微乳制劑的工業化發展提供借鑒。
1 儀器與試藥
1.1 儀器 Zetasizer Nano ZS90型納米粒徑電位分析儀(MALVERN公司);HT7700型透射式電子顯微鏡(HITACHI公司);MS603S型天平(美國梅特勒-托利多公司);二級反滲透純化水制水機(廣州萬冠制藥設備公司)。
1.2 試藥 廣藿香揮發油(批號:P905696-23)、肉豆蔻酸異丙酯(IPM)(批號:1811858)均購于上海麥克林生化科技有限公司;油酸(OA)(批號:20220120)、吐溫80(TW80)(批號:20211201)、聚乙二醇400(PEG400)(批號:20211215)、1,2-丙二醇(批號:20210901)、無水乙醇(批號:20211201)均購于天津大茂化學試劑廠;聚氧乙烯氫化蓖麻油(RH40)(批號:2022032038R)購于廣東翁江試劑公司;所有試劑均為分析純,藥用級別純化水等。
2 方法與結果
2.1 目測法和電導率-含水量曲線法制備空白微乳和廣藿香揮發油微乳 從預試驗篩選出3個穩定的空白微乳處方,分別制備3個空白微乳和3個載藥微乳。(見表1)(1)電導率-含水量曲線法:以處方3為例,將助表面活性劑、表面活性劑先混勻,再加入油相,在500 r/min轉速下磁力攪拌10 min,獲得4 g的微乳原液,然后加入0.2 g的純化水,攪拌20 s后,測電導率值并記錄,重復上述操作步驟,直至測得微乳含水量80%時電導率值,將得到的微乳含水量值、對應的電導率值,使用軟件Origin 2021繪制電導率-含水量曲線,將電導率獲得最大值時,對應的含水量值作為O/W型微乳成型臨界點。(見圖1)(2)目測法:將助表面活性劑、表面活性劑先混勻,再加入油相,500 r/min轉速下磁力攪拌10 min,獲得4 g的微乳原液,然后慢慢加入純化水,攪拌,觀察微乳狀態,重復上述操作,直至微乳由混濁狀態變為澄清,記錄加入純化水的質量,此時微乳的含水量值即為O/W型微乳成型臨界點。

圖1 處方3 空白微乳、處方6 載藥微乳的電導率-含水量曲線圖

表1 空白微乳和載藥微乳處方組成、配比
上述兩種配制方法,由3名操作人員單獨進行,將獲得的實驗結果取平均值,作為目測法和電導率-含水量曲線法的O/W型微乳成型臨界點,然后進一步比較兩種方法制備的微乳的平均粒徑和多分散系數(PDI)。由于單獨使用廣藿香揮發油作為油相取代處方1、2的OA、處方3的IPM,均不能獲得穩定的微乳(出現混濁、分層);將廣藿香揮發油與OA、廣藿香揮發油與IPM按質量比2∶1混勻后,作為混合油相,按表1處方1、2、3的配比,獲得微乳的平均粒徑、PDI偏大,穩定性一般;將廣藿香揮發油與OA、廣藿香揮發油與IPM按質量比1∶1混勻后,獲得微乳的平均粒徑、PDI均較小,穩定性較好,故選定該比例作為混合油相,備用。
2.2 目測法和電導率-含水量曲線法結果對比 依據“2.1”項表格的處方單獨進行,將獲得的實驗結果取平均值,作為目測法和電導率-含水量曲線法的O/W型微乳成型臨界點。各取兩種方法獲得的微乳適量,稀釋100倍后測量平均粒徑、PDI,實驗結果見表2。結果表明,目測法測得的O/W型微乳成型臨界點普遍靠前,在該臨界點下測得微乳的平均粒徑、PDI比電導率-含水量曲線法偏大,將處方1~6微乳各取適量,分別滴入亞甲基藍試劑和蘇丹Ⅲ試劑,靜置觀察,可見上述微乳中,亞甲基藍試劑的擴散速度均明顯大于蘇丹Ⅲ試劑,表明上述微乳類型均為O/W型。該實驗證實了目測法和電導率-含水量曲線法均能對O/W型微乳成型臨界點有較準確的判斷,但是電導率-含水量曲線法精確度更高,獲得的微乳更為穩定,證實了目測法確定的O/W型微乳成型臨界點,體系可能處于W/O型和O/W型雙連續相轉變為O/W型的最后階段[10-11],尚未完全轉變為O/W型。該結果也進一步驗證了電導率-含水量曲線法確定的臨界點比目測法更為精確。

表2 目測法、電導率-含水量曲線法檢測結果
2.3 工藝篩選
2.3.1 電導率-含水量曲線法制備工藝的設定 將廣藿香揮發油分別與OA、IPM按質量比1∶1的比例混勻,作為油相備用,選取預試驗中篩選出的RH40、TW80 作為表面活性劑,PEG400、1,2-丙二醇、無水乙醇作為助表面活性劑。選擇在室溫(25 ℃)下進行試驗,首先使用磁力攪拌器在500 r/min轉速下將表面活性劑和助表面活性劑按照固定質量配比Km值(Km值=表面活性劑/助表面活性劑)為2∶1混勻,再按(含藥油相/總表面活性劑)質量比分別為1∶9、2∶8、3∶7、4∶6、5∶5、6∶4、7∶3、8∶2、9∶1,在500 r/min轉速下攪拌10 min制成微乳原液4 g,然后加入0.2 g純化水,攪拌20 s,測電導率,再加入0.2 g純化水,攪拌,測電導率,直至體系含水量為80%時,將獲得的含水量、電導率值輸入Origin 2021繪圖,建立電導率-含水量曲線,將電導率最大時的含水量值作為O/W型微乳成型臨界點,對于不能成乳或者成乳后平均粒徑未在10~100 nm范圍的處方予以剔除;此外,部分微乳成乳后,靜置24 h又重新出現混濁,這類處方也予以剔除;最后,根據油相、總表面活性劑、水相的占比,使用Origin 2021軟件給合格的微乳處方繪制偽三元相圖。
2.3.1.1 油相、表面活性劑、助表面活性劑類型的選定 按“2.3.1”項的方法,將不同類型的油相、表面活性劑、助表面活性劑一一組合,按照固定配比Km為2∶1,混合油相、總表面活性劑質量比分別為1∶9、2∶8、3∶7、4∶6、5∶5、6∶4、7∶3、8∶2、9∶1,配制微乳,將合格的微乳處方繪制偽三元相圖。結果顯示,圖2D的微乳成乳區域最大,故選定廣藿香揮發油/IPM作為含藥油相,TW80作為表面活性劑、PEG400作為助表面活性劑,用于下階段Km值的篩選。(見圖2)
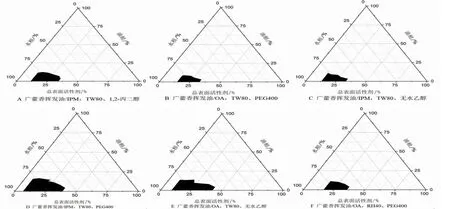
圖2 不同微乳處方的偽三元相圖
2.3.1.2 Km值的選定 按“2.3.1”項的方法,將微乳處方廣藿香揮發油/IPM,TW80、PEG400,選定Km為4∶1、3∶1、2∶1、1∶1,含藥油相、總表面活性劑質量比分別為1∶9、2∶8、3∶7、4∶6、6∶4、5∶5、6∶4、7∶3、8∶2、9∶1,配制微乳,篩選Km值。結果顯示,當Km值為3∶1時微乳成乳區域最大。(見圖3)

圖3 不同Km 值的偽三元相圖
2.3.2 單因素試驗“2.3.1.1”項和“2.3.1.2”項篩選結果表明,含藥油相與總表面活性劑質量比為1∶9、2∶8、3∶7、4∶6、5∶5、6∶4時,可獲得合格微乳,含藥油相與總表面活性劑質量比為7∶3、8∶2、9∶1時不能配制成乳,故選定含藥油相占比最小為10%,最大為60%(即廣藿香揮發油占比5%~30%)用于下一階段響應面法工藝篩選;“2.3.1.2”項篩選結果表明,表面活性劑與助表面活性劑質量配比Km值為2∶1、3∶1時可獲得較大的微乳成乳區域,當Km值為4∶1或1∶1時微乳成乳區域明顯變小,說明上述兩種質量配比下,不能獲得較大的含藥油相占比(微乳載藥量)。當Km值>4∶1或Km值<1∶1時,微乳成乳區域會進一步縮小,因此選定最大Km值為4∶1,最小Km值為1∶1進行下一階段響應面法工藝篩選。試驗過程中發現磁力攪拌器轉速的不同,對微乳的成乳效果有一定的影響,為了考察磁力攪拌器轉速對微乳成乳效果的影響,采取固定含藥油相與總表面活性劑質量比為1∶9、Km為3∶1,考察磁力攪拌器轉速對微乳成乳效果的影響。結果發現,磁力攪拌器轉速低于500r/min時成乳時間較長,且攪拌不夠均勻,不利于后續微乳的配制。當磁力攪拌器轉速高于1 100 r/min時,過高的轉速帶動了燒杯的轉動影響了攪拌效果,且過高的轉速有可能將空氣帶入,制得的微乳有氣泡分散在體系中。基于上述原因,選定磁力攪拌器轉速為500~1100 r/min。
2.3.3 響應面法工藝篩選[12-13]
2.3.3.1 響應面法考察因素 按“2.3.2”項的結果制定響應面法考察因素水平表。(見表3)

表3 響應面法考察因素水平表[14-15]
2.3.3.2 方案設計及結果[16]使用軟件Design-Expert 12.0對表3進行響應面法設計,統計結果,見表4。

表4 工藝和結果
對表4的獲得的結果進行平均粒徑、PDI的方差分析,見表5~6。結果顯示,平均粒徑、PDI獲得的數學模型均為極顯著性,且失擬項不具顯著性,表明上述數學模型模擬的情況較為準確[17-18]。顯著因素廣藿香揮發油(%)、Km對粒徑、PDI響應面分析見圖4。平均粒徑、PDI取最低值時擬合方程分別為:平均粒徑=27.230+5.790A+1.875B-0.426C+2.905AB-0.688AC+1.835BC-2.912A2+2.105B2+0.344AC2;PDI=0.102+0.037A+0.024B+0.005C+0.039AB+0.001AC+0.015BC-0.006A2+0.044B2-0.001C2。由于平均粒徑對微乳體系的影響更為重要,故選擇最小平均粒徑對應的點作為最優處方點,即廣藿香揮發 油 為10.07%、IPM 為10.07%、TW80 為60.43%、PEG400 為19.43%,Km為3.11,攪拌速率為622 r/min,獲得的平均粒徑理論值為17.64 nm,PDI為0.069。

圖4 廣藿香揮發油、Km 對平均粒徑、PDI 的響應面圖
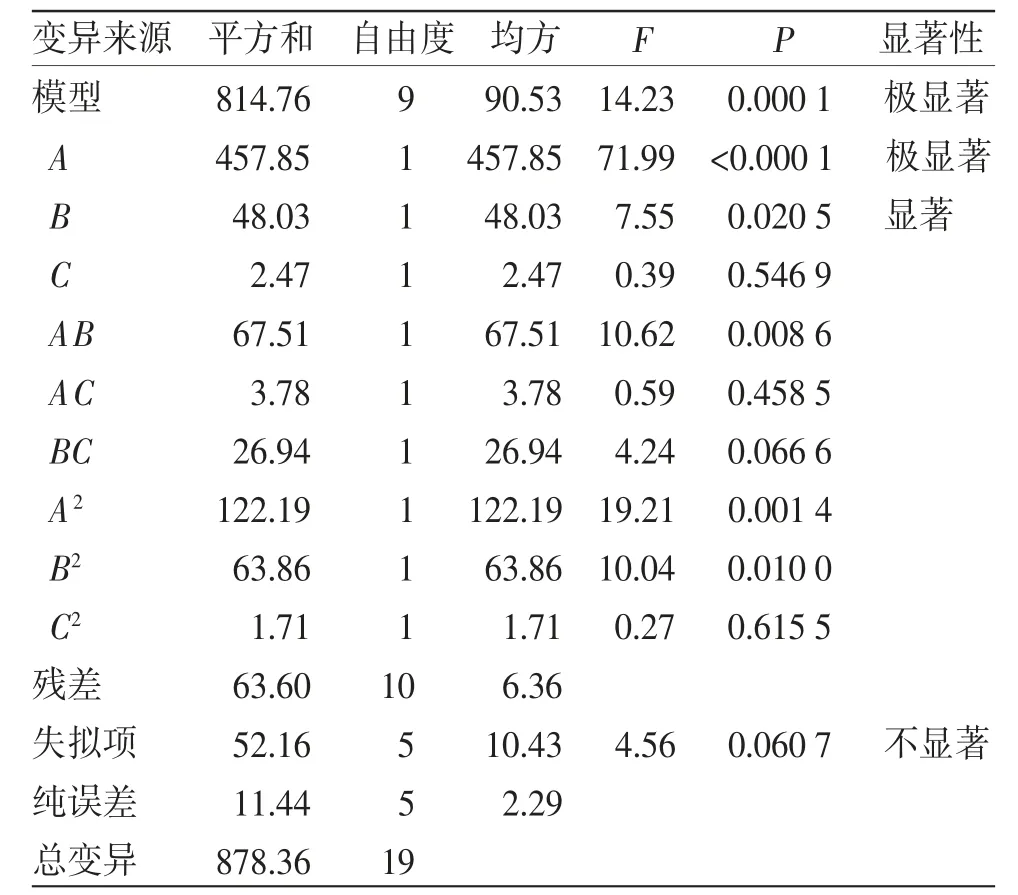
表5 平均粒徑的方差分析

表6 PDI 的方差分析
2.3.3.3 最優工藝驗證 對“2.3.3.2”獲得的最優點進行3批次驗證。結果顯示,平均粒徑、PDI值和模擬結果非常接近,說明此處方篩選結果可靠。(見表7)

表7 最優工藝驗證
2.4 微乳的理化性質及穩定性考察
2.4.1 微乳的電鏡圖 取“2.3.3.3”項微乳適量滴至銅網上,靜置10 min后,使用濾紙吸干,再加入2%的磷鎢酸負染10 min,再用濾紙吸干剩余液體,置于電鏡下觀察。圖片顯示微乳外觀飽滿、圓整,穩定性較好。(見圖5)

圖5 微乳的形態
2.4.2 高速離心考察 將微乳于5 000 r/min轉速下高速離心30 min。結果顯示,微乳依舊澄清、透明,未見分層,表明微乳穩定性好。
2.4.3 微乳類型鑒定及25 ℃下遮光、不遮光考察 取“2.3.3.3”項微乳適量,分別滴入亞甲基藍和蘇丹Ⅲ試劑,靜置5 min。圖6A、6B顯示,亞甲基藍在微乳中的擴散效果明顯,而蘇丹Ⅲ停留在液面幾乎不擴散,表明該微乳為O/W型,再取“2.3.3.3”項微乳適量,在室溫(25 ℃)條件下,分別作遮光處理和不作遮光處理,放置60 d后測平均粒徑、PDI,試驗平行3次。圖6C、6D顯示,兩種狀態下微乳的顏色無明顯差異;遮光狀態下,測得微乳的平均粒徑為(19.23±0.26)nm、PDI為(0.085±0.005);不遮光狀態下,測得微乳的平均粒徑為(19.47±0.31)nm、PDI為(0.089±0.009)。試驗結果進一步證明,在25 ℃下,微乳放置60 d后平均粒徑、PDI雖略為增大,但依舊穩定。

圖6 不同狀態下微乳的外觀
3 討 論
筆者查閱文獻[19]發現可使用滲濾電導模型理論解釋微乳的電導率-含水量曲線變化規律。以圖1為例,微乳原液階段,由于含水量為0,油的導電性又極低,此時電導率接近0。隨著含水量的增加,體系間連續相依舊為油相,故電導率的增加較為緩慢。當含水量到達滲濾閾值[20](滲濾閾值:導電微粒占整個體系的體積分數)后,帶電液滴增加明顯,故電導率增加迅速,此時體系為W/O型;隨著含水量的進一步增加,微乳液滴與液滴之間發生碰撞,形成了導電鏈,微乳的電導率又進一步增加,直至電導率的頂點,此范圍的微乳為W/O型和O/W型雙連續型。當微乳的電導率到達頂點后,含水量的增加又稀釋了帶電的微乳液滴,從而電導率開始下降,此時體系轉變為單一的O/W型。該理論可以有效解釋微乳電導率的變化與微乳的相間關系,但是之前的報道沒有采用直觀的方式來證實該理論的可靠性,而本研究則通過拍攝微觀狀態下微乳的形態驗證了該理論的可靠性。此外,試驗過程中發現攪拌速率對微乳的成乳有一定的影響,但是實際篩選結果卻并不顯著,因此,微乳的攪拌速率適中即可,太快可減少乳化時間,但容易帶入空氣,太慢則成乳時間過長,或難于配制成乳。再者,適當增加微乳的載藥量可減少服藥量,但是載藥量不宜一味增大,試驗過程中發現微乳載藥過大容易造成平均粒徑增大破壞其穩定性,甚至出現分層、變混濁的現象,因此,要擴大中藥微乳制劑,還須提高中藥精制、提純技術方可實現。本研究通過測定平均粒徑、PDI及生物透射電鏡觀察的手段驗證了電導率-含水量曲線法中電導率最大值作為O/W型微乳成型臨界點的精確性,可以為中藥微乳制劑的制備提供一定的參考。
