小分子c-Met激酶抑制劑Foretinib的合成
2023-09-22 05:55:14趙培培
合成化學 2023年9期
趙培培, 張 帆, 王 剛
(錦州醫科大學 藥學院,遼寧 錦州 121001)
Foretinib(XL880),化學名為N-{3-氟-4-{6-甲氧基-7-[3-(4-嗎啉)丙氧基]喹啉}-4-氧基-苯基}-N-(4-氟苯基)-2,2-二亞甲基丙二酰胺,是由美國Exelixis公司研發的一種對肝細胞生長因子(hepatocyte growth factor, HGF)和血管內皮生長因子受體(vascular endothelial growth factor receptor, VEGFR)具有雙重抑制作用的小分子多靶點酪氨酸激酶抑制劑[1]。體外研究表明:Foretinib對Met和KDR的抑制作用最強,IC50值分別為0.40 nmol/L和0.86 nmol/L;對Ron、 KIT、 Flt-1/3/4、 PDGFR-β和Tie-2也具有特殊的抑制活性,但作用稍弱,對Ron、 KIT、 Flt-1、 Flt-3和Flt-4的IC50值分別為3.00 nmol/L、 6.70 nmol/L、 6.80 nmol/L、 3.60 nmol/L和2.80 nmol/L;對成纖維生長因子受體(fibroblast growth factor receptors, FGFR)和表皮生長因子受體(epidermal growth factor receptor, EGFR)幾乎沒有抑制活性[2-3]。目前,Foretinib對于頭頸部癌、乳頭狀腎癌、肝細胞癌、胃癌和非小細胞肺癌等腫瘤的治療處于不同的臨床試驗階段[4-11]。此外,尚有臨床前研究表明,Foretinib對食管腺癌、人膠質母細胞癌和卵巢癌等惡性腫瘤細胞具有顯著的抑制作用[1, 12-13]。
目前,已有多篇文獻報道了Foretinib的合成路線。文獻[14]是以4-(3-鹵代丙氧基)-3-甲氧基苯乙酮為原料,經硝化、與嗎啉親核取代、硝基還原、與甲酸乙酯環合、氯代、與2-氟-4-硝基苯酚醚化、硝基還原和與1-[(4-氟苯基)胺甲酰基]環丙烷-1-羧酸酰化反應制得目標化合物。文獻[15]是以4′-羥基-3′-甲氧基苯乙酮(2)為原料,經與N-氯丙基嗎啉醚化、硝化、硝基還原、與甲酸乙酯環合、與3,4-二氟硝基苯醚化、硝基還原以及與1-[(4-氟苯基)胺甲酰基]環丙烷-1-羧酸(2)酰化反應制得目標化合物,總收率為0.76%。與文獻[15]相比,文獻[16]僅調整了反應順序,4′-羥基-3′-甲氧基苯乙酮先與1-溴-3-氯丙烷發生醚化反應,再進行硝化和嗎啉親核取代反應制得中間體5-甲氧基-4-[3-(4-嗎啉基)丙基]氧基-2-硝基苯乙酮(5),后續反應步驟與文獻[15]基本相同,總收率為4.56%。上述路線普遍存在原輔料價格昂貴、反應體系復雜、所用試劑種類較多、操作繁瑣及收率低等缺點,不適合工業化生產。本文在參考已有合成路線的基礎上,對Foretinib的合成路線和合成工藝進行了優化,路線見圖1,以4′-羥基-3′-甲氧基苯乙酮為起始原料,經與1-溴-3-氯丙烷醚化、硝化、嗎啉親核取代、與N,N-二甲基甲酰胺二甲基縮醛縮合、還原環合、氯代和與2-氟-4-硝基苯酚醚化和硝基還原制得氨基化合物(10);另一方面,1,1-環丙二羧酸氯代后與4-氟苯胺反應生成化合物(12);最后,化合物10和化合物12發生酰化反應制得目標化合物Foretinib(1),反應總收率為26.06%,目標化合物純度為99.40%。該路線原輔料價廉易購得,反應條件溫和,操作及后處理簡單,與已有路線相比總收率提高了5.71~34.29倍,更適合于大規模生產。
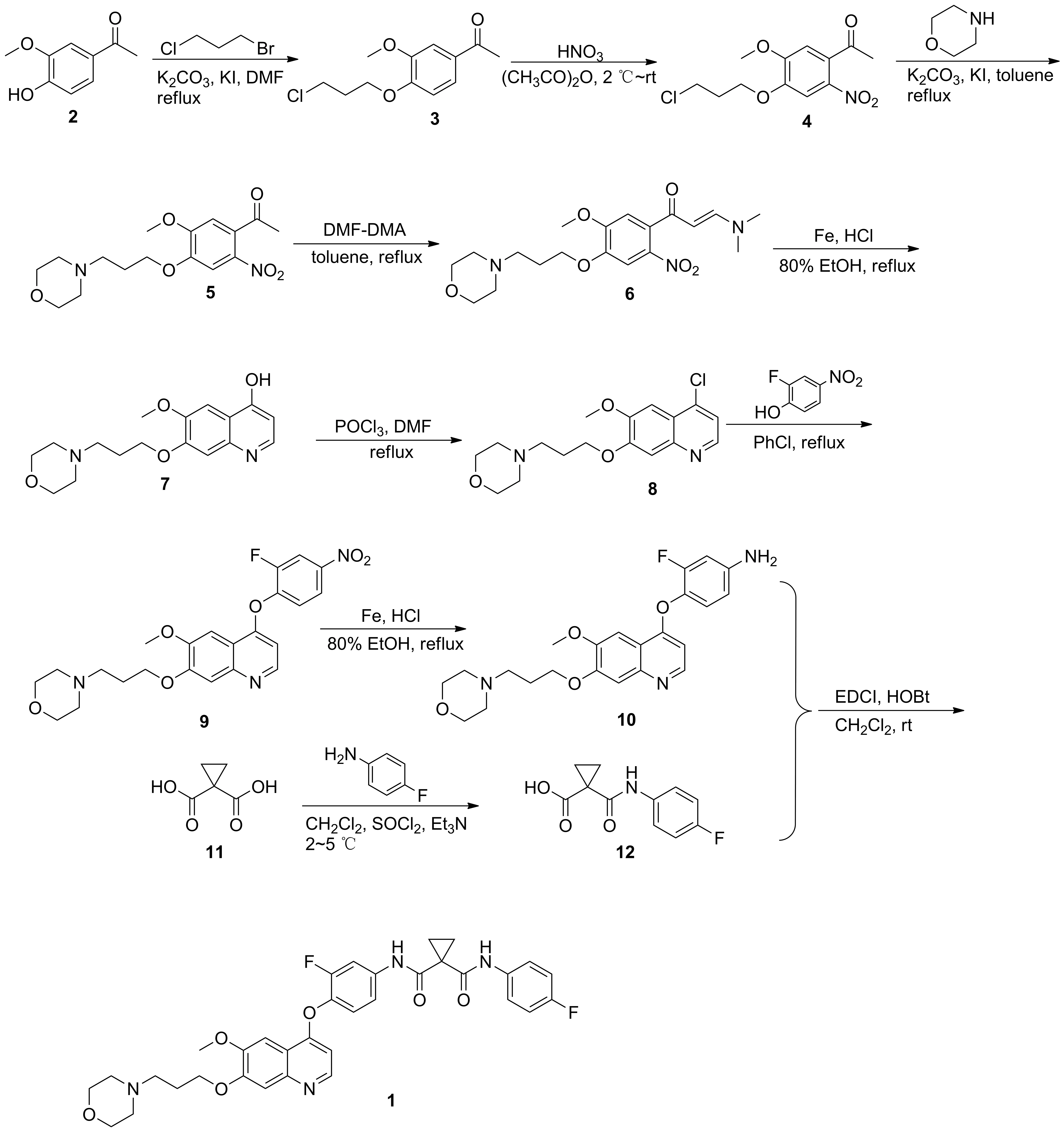
圖1 Foretinib的合成路線
1 實驗部分
1.1 儀器與試劑
MP50型超越熔點儀(溫度未經校正);AVANCE III 400 MHz型核磁共振儀(CDCl3和DMSO-d6為溶劑,TMS為內標);TSQ質譜儀;日立L-2000型高相液相色譜儀。
所用試劑均為市售分析純或化學純。
1.2 合成
(1) 4-(3-氯丙氧基)-3-甲氧基苯乙酮(3)的合成
將5.00 g(30.09 mmol)4′-羥基-3′-甲氧基苯乙酮(2)溶解于10.00 mL的N,N-二甲基甲酰胺中,依次加入5.82 g碳酸鉀(42.11 mmol)和0.25 g(1.51 mmol)碘化鉀,室溫攪拌30 min后,向上述反應液中滴加4.15 mL(42.09 mmol)1-溴-3-氯丙烷,繼續室溫反應8 h。在劇烈攪拌下,將反應液緩慢倒入冰水中,攪拌30 min,抽濾,用水洗滌濾餅,干燥,得白色固體7.11 g,收率97.41%, m.p.61.9~63.0 ℃;1H NMR(CDCl3, 400 MHz),δ: 7.56(d,J=5.6 Hz, 1H), 7.53(s, 1H), 6.92(d,J=5.6 Hz, 1H), 4.24(t,J=4.0 Hz, 2H), 3.92(s, 3H), 3.78(t,J=4.0 Hz, 2H), 2.57(s, 3H), 2.35~2.31(m, 2H)。
(2) 4-(3-氯丙氧基)-5-甲氧基-2-硝基苯乙酮(4)的合成
將5.00 g(20.60 mmol)化合物3溶解于10.00 mL乙酸酐中,將反應瓶置于冰浴中,控制反應溫度≤2 ℃,在此溫度下向反應液中緩慢滴加2.00 mL(28.89 mmol)濃硝酸,滴加過程控制溫度≤10 ℃,滴加完畢,升溫至室溫反應30 min。在劇烈攪拌下,將上述反應液快速倒入冰水中,攪拌1 h,抽濾,干燥,得粗品,70.00%乙醇重結晶,得黃色針狀晶體2.86 g,收率48.25%, m.p.67.3~69.2 ℃;1H NMR(CDCl3, 400 MHz),δ: 7.65(s, 1H), 6.76(s, 1H), 4.26(t,J=4.0 Hz, 2H), 3.96(s, 3H), 3.78(t,J=4.0 Hz, 2H), 2.50(s, 3H), 2.36~2.32(m, 2H)。
(3) 5-甲氧基-4-[3-(4-嗎啉基)丙基]氧基-2-硝基苯乙酮(5)的合成
將5.00 g(17.38 mmol)化合物4溶解于20.00 mL甲苯中,然后依次加入3.60 g(26.05 mmol)碳酸鉀和0.15 g(0.90 mmol)碘化鉀,室溫攪拌30 min后,向上述反應液中滴加6.06 mL(69.56 mmol)嗎啉,滴加完畢,升溫至回流反應13 h。待反應液冷卻至室溫后,減壓濃縮反應液至干,向殘留物中加入25.00 mL水,室溫攪拌30 min,抽濾,用水洗滌濾餅,干燥,得黃色固體5.78 g,收率98.30%, m.p.93.8~94.7℃;1H NMR(CDCl3, 400 MHz),δ: 7.64(s, 1H), 6.75(s, 1H), 4.19(t,J=4.4 Hz, 2H), 3.96(s, 3H), 3.72(t,J=3.2 Hz, 4H), 2.53(t,J=4.4 Hz, 2H), 2.50(s, 3H), 2.47(s, 4H), 2.08~2.04(m, 2H); ESI-MS,m/z: 339.15{[M+H]+}。
(4) 1-{5-甲氧基-4-[3-(4-嗎啉基)丙氧基]-2-硝基}苯基-3-二甲氨基-2-烯-1-丙酮(6)的合成
將5.00 g(14.78 mmol)化合物5和10.80 mL(81.30 mmol)N,N-二甲基甲酰胺二甲基縮醛依次加入到15.00 mL甲苯中,回流反應7 h。待反應液冷卻至室溫后刮壁,有固體析出,抽濾,用甲苯洗滌濾餅,干燥,得金黃色固體5.63 g,收率96.84%, m.p.149.7~150.5 ℃;1H NMR(CDCl3, 400 MHz),δ: 7.61(s, 1H), 6.84(s, 1H), 5.20(s, 1H), 4.17(t,J=4.4 Hz, 2H), 3.94(s, 3H), 3.73(t,J=3.2 Hz, 4H), 3.08(s, 3H), 2.85(s, 3H), 2.53(t,J=4.4 Hz, 2H), 2.47(s, 4H), 2.07~2.03(m, 2H); ESI-MS,m/z: 394.20{[M+H]+}。
(5) 6-甲氧基-7-[3-(4-嗎啉基)丙氧基]-4-羥基喹啉(7)的合成
將4.27 g還原鐵粉(76.25 mmol)加入到25.00 mL的80%乙醇中,滴加10滴濃鹽酸,回流反應30 min。稍冷,向上述反應液中分批加入5.00 g(12.71 mmol)化合物6,繼續回流反應2 h。趁熱抽濾,濾餅用95.00%乙醇繼續回流20 min后,趁熱抽濾,重復2次。合并濾液,減壓濃縮濾液至干。向殘留物中加入50.00 mL水,用飽和碳酸鈉溶液調節pH至9,抽濾,濾餅用水打漿,抽濾,用水洗滌濾餅,干燥,得黃綠色固體3.60 g,收率88.98%, m.p.84.2~86.2 ℃;1H NMR(CDCl3, 400 MHz),δ: 11.48(s, 1H), 7.70(s, 1H), 7.61(d,J=4.4 Hz, 1H), 6.98(s, 1H), 6.24(d,J=4.8 Hz, 1H), 4.09(t,J=4.0 Hz, 2H), 3.91(s, 3H), 3.68(t,J=2.8 Hz, 4H), 2.49(t,J=4.8 Hz, 2H), 2.43(s, 4H), 2.06~2.03(m, 2H); ESI-MS,m/z: 319.17{[M+H]+}。
(6) 4-氯-6-甲氧基-7-[3-(4-嗎啉基)丙氧基]喹啉(8)的合成
將5.00 g(15.70 mmol)化合物7分批加入到25.00 mL三氯氧磷中,滴加3滴N,N-二甲基甲酰胺,回流反應1 h。反應液冷卻至室溫后,減壓濃縮反應液至干,在劇烈攪拌下,將殘留物倒入碎冰中,用飽和碳酸鈉溶液調節pH至9,抽濾,濾餅用水打漿,干燥,得灰白色固體4.94 g,收率93.39%, m.p.107.4~108.2 ℃;1H NMR(CDCl3, 400 MHz),δ: 8.57(d,J=4.8 Hz, 1H), 7.44(s, 1H), 7.40(s, 1H), 7.35(d,J=4.8 Hz, 1H), 4.27(t,J=6.8 Hz, 2H), 4.04(s, 3H), 3.73(t,J=4.4 Hz, 4H), 2.57(t,J=6.8 Hz, 2H), 2.48(s, 4H), 2.16~2.09(m, 2H); ESI-MS,m/z: 337.13{[M+H]+}。
(7) 6-甲氧基-7-[3-(4-嗎啉基)丙氧基]-4-(2-氟-4-硝基苯基)喹啉(9)的合成
將5.00 g(14.84 mmol)化合物8和3.03 g(19.29 mmol)的2-氟-4-硝基苯酚依次加入到25.00 mL氯苯中,回流反應6 h,待反應液冷卻至室溫后,減壓濃縮反應液至干,向殘留物中加入50.00 mL水,用1.00 mol/L氫氧化鈉溶液調節pH至10,室溫攪拌1 h,抽濾,濾餅用水打漿,干燥,得淡黃色固體6.07 g,收率89.38%, m.p.135.4~137.5 ℃;1H NMR(CDCl3, 400 MHz),δ: 8.58(d,J=5.2 Hz, 1H), 8.19(dd,J=9.6 Hz, 2.4 Hz, 1H), 8.15~8.12(m, 1H), 7.48(s, 1H), 7.43(s, 1H), 7.34(t,J=8.0 Hz, 1H), 6.55(d,J=5.2 Hz, 1H), 4.29(t,J=6.4 Hz, 2H), 4.02(s, 3H), 3.73(t,J=4.4 Hz, 4H), 2.58(t,J=7.2 Hz, 2H), 2.49(s, 4H), 2.18~2.11(m, 2H); ESI-MS,m/z: 458.17{[M+H]+}。
(8) 6-甲氧基-7-[3-(4-嗎啉基)丙氧基]-4-(2-氟-4-氨基苯基)喹啉(10)的合成
將2.45 g(43.72 mmol)還原鐵粉加入到25.00 mL的95%乙醇中,滴加濃鹽酸10滴,回流反應30 min。稍冷,向上述反應液中分批加入5.00 g(10.93 mmol)化合物9,回流反應1 h。趁熱抽濾,濾餅用95.00%乙醇繼續回流20 min后,趁熱抽濾,重復2次。合并濾液,待濾液冷卻至室溫,抽濾,干燥,得淺棕色固體。濾液減壓濃縮至干,向殘留物中加入20.00 mL水,用飽和碳酸鈉溶液調節pH至8,抽濾,干燥,得淺粉色固體,合并2次所得固體,共4.29 g,收率91.82%, m.p.170.5~171.5 ℃;1H NMR(CDCl3, 400 MHz),δ: 8.47(d, 1H,J=5.2 Hz), 7.58(s, 1H), 7.42(s, 1H), 7.04(t,J=8.4 Hz, 1H), 6.56(d,J=11.6 Hz, 1H), 6.51(d,J=8.4 Hz, 1H), 6.40(d,J=4.8 Hz, 1H), 4.27(t,J=6.4 Hz, 2H), 4.04(s, 3H), 3.83(s, 2H), 3.73(s, 4H), 2.58(t,J=6.8 Hz, 2H), 2.49(s, 4H), 2.15~2.12(m, 2H); ESI-MS,m/z: 428.20{[M+H]+}。
(9) 1-[(4-氟苯基)胺甲酰基]環丙烷-1-羧酸(12)的合成
將5.00 g(38.43 mmol)1,1-環丙二羧酸(11)加入到25.00 mL二氯甲烷中,將反應瓶置于冰浴中,控制溫度≤2 ℃,在此溫度下向反應液中緩慢滴加5.86 mL(42.27 mmol)三乙胺,滴加過程控制溫度≤5 ℃,滴加完畢,冰浴反應30 min。向上述反應液中緩慢滴加3.07 mL(42.27 mmol)氯化亞砜,滴加過程控制溫度≤5 ℃,滴加完畢,冰浴反應2 h。再向反應液中緩慢滴加4.00 mL(42.27 mmol)4-氟苯胺,滴加完畢,冰浴反應3 h。向上述反應液中加入50.00 mL的10%(w/v)氫氧化鈉溶液,分離有機相,用10%(w/v)氫氧化鈉溶液萃取(3×10.00 mL),棄去有機相。合并水相,
用二氯甲烷(2×10.00 mL)洗滌。水相用2.00 mol/L鹽酸溶液調節pH至2~3,抽濾,干燥,得白色固體5.62 g,收率65.52%, m.p.181.8~184.4 ℃;1H NMR(DMSO-d6, 400 MHz),δ: 10.66(s, 1H), 7.61(dd,J=5.6 Hz, 2.4 Hz, 2H), 7.15(t,J=6.0 Hz, 2H), 1.41(s, 4H); ESI-MS,m/z: 222.06{[M-H]-}。
(10) Foretinib(1)的合成
將3.13 g(14.02 mmol)化合物12、 3.13 g(23.37 mmol)1-乙基-(3-二甲基氨基丙基)碳二亞胺鹽酸鹽和2.37 g(17.54 mmol)1-羥基苯并三唑一水合物加入到25.00 mL二氯甲烷中,室溫反應30 min,然后將5.00 g(11.70 mmol)化合物10加入上述反應液中,室溫反應12 h。反應完畢,抽濾,棄去濾餅,濾液用50.00 mL水稀釋,室溫攪拌1 h,分離有機相,水相用二氯甲烷萃取(3×10.00 mL),合并有機相,依次用飽和碳酸氫鈉溶液(3×10.00 mL)洗滌,水(3×10.00 mL)洗滌,無水硫酸鈉干燥,減壓蒸除溶劑,干燥,得淡黃色固體6.32 g,收率85.41%, m.p.99.3~101.1 ℃;1H NMR(DMSO-d6, 400 MHz),δ: 10.40(s, 1H), 10.01(s, 1H), 8.47(d,J=6.8 Hz, 1H), 7.93~7.88(m, 1H), 7.65(dd,J=11.6 Hz, 6.8 Hz, 2H), 7.53(s, 2H), 7.45~7.39(m, 2H), 7.16(t,J=11.6 Hz, 2H), 6.42(d,J=6.8 Hz, 1H), 4.20(t,J=8.0 Hz, 2H), 3.95(s, 3H), 3.60(t,J=5.6 Hz, 4H), 2.51~2.41(m, 6H), 2.01~1.96(m, 2H), 1.48(s, 4H); ESI-MS,m/z: 633.25{[M+H]+}; 純度99.40%[色譜柱:Venusil XBP C18, 4.6×250 mm×5 μm,柱溫:30 ℃,檢測波長:241 nm,流動相:乙腈∶醋酸銨溶液(pH=5.2)=50 ∶50,V∶V]。
2 結果與討論
2.1 化合物4的合成
化合物3的結構中2-位、3-位和6-位均可發生硝化反應,但反應難易程度不同。為了得到2-位的單硝化產物,從反應收率、產物純度和操作是否簡便等角度出發,本文分別考察了不同的反應溶劑、硝化試劑及反應溫度對硝化反應的影響,結果如表1所示。當以乙酸酐為反應溶劑,濃硝酸為硝化試劑25 ℃反應時,反應時間短,僅為0.5 h,收率較高。且反應結束后,將反應液倒入冰水中可直接得到固體產物,無需萃取,操作簡便。本文還對濃硝酸的用量進行了優化,通過TLC監測發現,當濃硝酸的用量為1.1 eq.時,即使延長反應時間仍有原料剩余;當濃硝酸的用量≥1.2 eq.時,化合物3可以全部轉化為化合物4。其中,濃硝酸用量為1.4 eq.,收率最高可達48.25%。
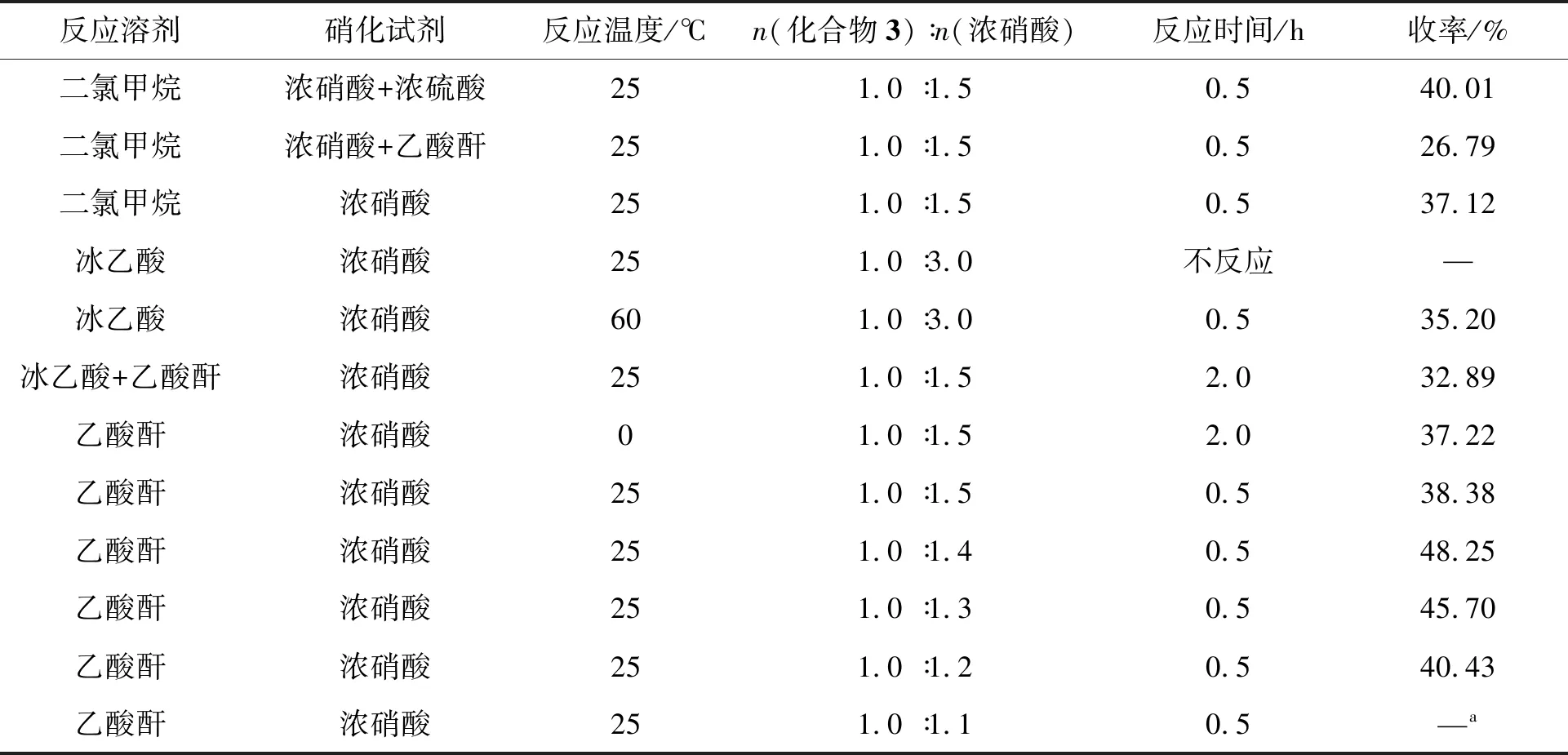
表1 硝化試劑、反應溶劑和反應溫度對硝化反應結果的影響
2.2 化合物7的合成
文獻[14-16]報道的合成4-羥基喹啉化合物7的方法一般是:硝基化合物5在鈀/碳或鐵/氯化銨作用下還原為氨基,然后再與甲酸乙酯在醇鈉的作用下環合形成喹啉環。文獻[17]制備喹啉環時,選擇鄰硝基苯乙酮化合物為原料,首先羰基α-位的甲基與N,N-二甲基甲酰胺二甲基縮醛發生縮合反應得到1-苯基-3-二甲氨基-2-烯-1-丙酮化合物,然后該化合物結構中的硝基被還原為氨基后,發生自身環合反應形成喹啉環。本文采用上述方法由化合物5經與N,N-二甲基甲酰胺二甲基縮醛縮合和還原環合2步反應合成得到化合物7,反應歷程見圖2, 2步反應總收率可達86.17%,比文獻[15-16]分別提高了30.81%、 24.82%,而且該方法反應條件溫和,使用試劑種類更少。

圖2 化合物7的合成機理
2.3 化合物9的合成
化合物8與2-氟-4-硝基苯酚反應制備化合物9為醚化反應。醚化反應一般是在非質子性溶劑中,有機堿或無機堿的存在下進行的。2-氟-4-硝基苯酚結構中的硝基和氟原子都是吸電子基團,使氧原子的電子云密度降低,親核性下降,酚羥基的親核取代反應變得困難,因此文獻[14-16]中使用了堿性較強的碳酸銫和2,6-二甲基吡啶。由于反應物和產物在高溫條件下都比較穩定,可以通過升高反應溫度加速反應的進行,分別嘗試了N,N-二甲基甲酰胺、二甲基亞砜、二苯醚、二甲苯和氯苯作為反應溶劑,在140 ℃左右反應。研究表明,當以氯苯和二甲苯為反應溶劑時,無需堿的參與,醚化反應也可順利進行,而且反應收率較高,均大于85.00%,從生產成本考慮,選擇價格較為便宜的氯苯作為醚化反應溶劑。
2.4 化合物1的合成
化合物10與化合物12反應生成化合物1的本質是芳氨基的酰化反應。上述酰化反應可以由羧基化合物12首先經氯代反應轉化為酰氯,再與氨基化合物10在三乙胺存在下發生酰化反應。通過TLC監測發現,該方法會在酰化過程中產生多個雜質,且極性與目標化合物相近,增加了分離純化的難度。將三乙胺更換為4-二甲氨基吡啶也不能達到理想的效果。酰化反應也可以由氨基化合物10和羧基化合物12在多肽縮合劑的作用下直接反應。本文嘗試了由與化合物10和化合物12在縮合劑1-乙基-(3-二甲基氨基丙基)碳二亞胺鹽酸鹽和催化劑1-羥基苯并三唑一水合物的作用下制備目標化合物1。通過TLC監測發現,該方法沒有雜質生成,并且1-乙基-(3-二甲基氨基丙基)碳二亞胺鹽酸鹽和1-羥基苯并三唑一水合物水溶性較好,易于除去,收率和目標化合物的純度都較高。
3 結論
本文設計的Foretinib的合成路線選擇價廉易得的4′-羥基3′-甲氧基苯乙酮(2)作為起始原料,經過醚化、硝化、親核取代、縮合、還原環合和氯代等9步反應制備得到Foretinib(1),總收率為26.06%,純度為99.40%。本合成路線反應條件溫和,操作簡單,后處理簡便,各步反應收率均較高,為Foretinib的上市銷售和工業化生產奠定了基礎。
