Mazabraud 綜合征的臨床、影像學及病理學特征:1 例報道暨文獻復習
2023-10-07 13:13:52沈艷楊通陳貴東古婉儀
腫瘤預防與治療 2023年9期
沈艷,楊通,陳貴東,古婉儀
510260 廣州,廣州醫科大學附屬第二醫院 病理科
Mazabraud 綜合征是以單骨性或多骨性的纖維結構不良(fibrous dysplasia,FD)合并單發或多發性肌內黏液瘤(intramuscular myxoma,IM)為臨床特征的罕見疾病[1],發病率約1/1 000 000(www.orpha.net)。由于該疾病常缺乏明顯的臨床癥狀,并且臨床工作者認識不足易導致漏診,預計其發病率可能高于預期。 鑒于骨病變惡性轉化的風險增加,有必要提高對Mazabraud 綜合征的認識,避免誤診、漏診。現報道我院收治的1 例Mazabraud 綜合征,并復習相關文獻,探討其病因、臨床特點和病理學特征,以期加深臨床醫師對該疾病的了解。
1 病例報道
患者,女性,57 歲,因左臀部腫物3 年余伴左髖部疼痛1 月,于2021 年6 月入院。自述平素身體良好,否認既往有外傷史或腫瘤史。入院后彩超顯示左大腿根部內側皮下約3.8 mm 處脂肪層大小約66 mm× 23 mm× 39 mm 混合回聲團塊,邊界尚清晰,形態欠規則,內部回聲不均,可見液性暗區,后方回聲增強。X 線檢查顯示左側髂骨、左側股骨上段骨質密度不均勻,見斑片狀低密度影及稍高密度影(圖1A)。MRI 示左側股骨上段、髖臼、坐骨、髂骨見多發片狀異常信號,為T1WI 低信號混雜點狀高信號,T2WI 及壓脂序列混雜高信號,DWI 高信號,增強掃描可見不均勻強化(圖1B)。左側大腿內側軟組織見腫塊影,范圍約4.2 cm×2.8 cm,為T1WI 低信號、T2WI 高信號、壓脂序列高信號,增強掃描為不均勻增強,邊緣見環形強化(圖1C、D)。
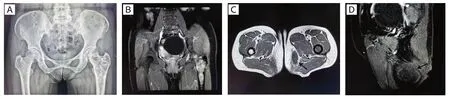
圖1 本病例的影像學表現Figure 1.Imaging Features of Present Case
完善相關檢查后,行左臀部腫物切除術及左股骨近端骨病灶刮除及植骨術。切除和刮除組織分別送病理檢查。大體觀察見:(1)左臀部腫物,大體為橢圓形,表面光滑,似有包膜,附著少量脂肪,大小5.5 cm×4.0 cm ×2.8 cm,切面呈實性,灰白色,質軟,有光澤,呈膠凍樣(圖2);(2)左股骨近端骨病灶刮除標本:碎組織一堆,約4.5 cm×4 cm×1.5 cm,灰白色,質較韌,刀切有砂礫感。鏡下觀察見:(1)左臀部腫物:腫瘤細胞稀疏,間質富含黏液樣基質,可見疏松的網狀纖維,血管稀少(圖3A)。細胞小且大小較一致,胞漿少,嗜酸性,呈梭形或星芒狀,核呈圓形或梭形,固縮深染,無異型性,無核分裂像(圖3B)。腫瘤周圍可見不完整的假性纖維包膜包繞,可見腫瘤細胞侵入脂肪間,與周邊肪組織分界不清(圖3C);(2)左股骨近端病灶:由增生的纖維樣組織和不成熟的編織狀新生骨構成,兩者比例在不同區域多少不等。纖維樣組織主要由纖維母細胞樣梭形細胞構成,形態溫和,無明顯異型性,核分裂少見;編織狀新生骨主要為纖細、形態不規則、彎曲的的骨小梁,呈“C””J”等英文字母形,骨小梁之間缺乏彼此連接,表面未見明顯骨母細胞襯覆(圖3D)。局部間質見黏液樣變及散在或灶性泡沫樣組織細胞浸潤。免疫組織化學染色發現,左臀部腫瘤細胞呈vimentin 陽性,Desmin、S-100、SMA、CD34、MUC4 均陰性。結合相關結果,本病例最終診斷為:Mazabraud 綜合征(左股骨近端FD 合并左臀部IM)。患者術后門診隨訪1 年10 個月,未見復發或惡性轉化。

圖2 軟組織腫物大體標本Figure 2.Gross Specimen of Soft Tissue Tumor

圖3 本病例的病理學特征Figure 3.Pathological Features of Present Case
2 討 論
1926 年Henschen[1]首次報道了1 例FD 同時伴發周圍多發性IM 的病例,1957 年Mazabraud 進一步詳細闡述了FD 與IM 之間的病理相關性,并以自己的名字將其命名為Mazabraud 綜合征[2]。這是一種罕見的散發性疾病,病因尚不明確。近年來,分子遺傳學研究顯示FD 和IM 都與GNAS1 基因突變有關[3-4],且突變熱點幾乎均位于第8 外顯子,提示GNAS1 基因突變在Mazabraud 綜合征發病中起著決定性作用。人G 蛋白分為激活型(Gs)和抑制型(Gi)兩種,由α、β、γ亞基組成,在細胞跨膜信號傳遞中起重要作用。α亞基是其活性亞基,含有GTP和 GDP 結合位點并具有GTP 酶活性[5]。GNAS1 基因突變可導致其編碼蛋白Gsa 的GTP 酶活性喪失,引起腺苷酸環化酶異常激活,繼而導致細胞內第二信號cAMP 的過表達,最終引起間充質細胞的前體細胞異常增殖[6-7],導致FD 和IM。由于GNAS1 基因突變發生在體細胞,因此病變組織中突變細胞和正常細胞呈鑲嵌分布,而其突變發生的時段和突變細胞的位置、比例決定了發病年齡、病變的范圍和嚴重程度。這從分子機制方面詮釋了Mazabraud 綜合征臨床表現的異質性。目前FD 和IM 的GNAS1基因突變研究多為非Mazabraud 綜合征病例,而Mazabraud 綜合征極為罕見,迄今對其GNAS1 基因突變研究的文獻報道十分有限。國內唐娟等[8]曾研究了1 例Mazabraud 綜合征的GNAS1 基因突變情況,在其FD 中發現了該基因的熱點突變,即第8 外顯子中的第201 位密碼子突變為CGT →TGT,但在同一患者的IM 中未檢測到突變,究其原因,可能因基因突變細胞太少而導致假陰性結果。Cox 等[9]對其報道的2 例Mazabraud 綜合征的其中1 例進行了GNAS1 基因突變分析,發現FD 和IM 均發生了相同的熱點突變,即在第201 位密碼子均發生了R201H突變。歐洲一項多中心研究對32 例Mazabraud 綜合征的流行病學及臨床特點進行了回顧性分析,其中在6 例行GNAS1 基因突變檢測的IM 中,發現5 例均存在第8 外顯子的熱點突變,但在這些患者的FD 病變中,因無標本可獲取而未能確定FD 病變的突變是否與IM 的突變相匹配[10]。最近,Ka?par 等[11]對其報道的1 例Mazabraud 綜合征進行了GNAS1 基因突變分析,在患者的FD 和IM 組織中均檢測到了R201H熱點突變。這些研究結果進一步證實了GNAS1 基因突變是Mazabraud 綜合征的重要病因機制。
Mazabraud 綜合征好發于女性,男∶女為1∶2。FD 中位發病年齡為40 歲。根據Vescini 等[12]對Mazabraud 綜合征的回顧性研究和文獻復習,100 例骨病變中,77 例為多骨性損傷(占77%),常見于股骨和盆骨,也可發生在脛骨、肋骨、顱骨等部位;在單側和單骨性損傷中,右側較左側更多見(57% vs 43%)。IM 發病中位年齡為47 歲,平均比FD 晚6.5年發病,也可單發或多發, 以多發為主(占60%),右側較左側更多見(61% vs 39%)。最常見于四肢、肩和臀部肌肉內,尤其大腿最多見,占一半以上。少數情況下,IM 還可見發生在頭頸部、胸壁、腹膜后、皮下及關節附近[13]。Hagelstein-Rotman 等[14]報道了來自荷蘭、美國和法國三級轉診中心就診的30 例Mazabraud 綜合征患者,發病中位年齡為42 歲,女性20 例(67%),男性10 例(33%),其中26 例患者被診斷為多骨性疾病(87%),29 例患者的IM 位于骨性FD 病變附近(97%)。趙紅葉等[15]于2008 年報道了國內首例Mazabraud 綜合征,該患者為女性,60 歲,以右大腿腘窩腫塊為首發癥狀,臨床表現為右側股二頭肌內近腘窩處單發IM 及同側股骨上段單骨性FD。其后,國內先后報道了數例Mazabraud 綜合征,均為個案報道,年齡從23 歲至70 歲不等,無明顯性別差異,全部病例均以軟組織腫塊為首發癥狀,多數病例IM 為單發,骨病變中所有病例均累及股骨,約一半病例為多發性骨損傷,左右側比例基本相同[16-18]。上述臨床特點與文獻報道有出入,可能與國內病例報道太少而造成的偏差有關。我們報道的這例為57 歲女性患者,FD 表現為多骨型,病變累及左側股骨上段、髖臼、坐骨及髂骨,即骨性病變見于左側股骨和左側盆骨;IM 發生在大腿根部,也是左側,貼近坐骨結節,臨床誤認為是坐骨結節囊腫。值得一提的是,解剖學上,本例IM 位于皮下脂肪深層,術中見腫瘤一側緊貼深筋膜(肌筋膜),而并非發生在肌肉內。根據發現左臀部腫物3 年余的病史,推測IM 發病至少在4 年前;而FD 臨床主要表現為近期骨疼,由于之前無手術史、骨折史且無任何骨骼影像學檢查,因此無法推測FD 發生的大概年齡及是否早于IM 發病。
Mazabraud 綜合征中的FD 其臨床癥狀與病變部位和骨損范圍、程度有關,可表現為局部膨隆和腫塊、疼痛、畸形等。影像學上,通常表現為界限清楚的低密度膨脹性溶骨性病變,病灶周圍骨皮質厚薄不一,邊緣有致密硬化,無骨膜反應[19]。FD 典型X 線表現為毛玻璃樣外觀,周圍有致密的骨質,邊界清晰,皮質通常完整,但由于膨脹性病變而可能變薄;如果存在軟骨島,在X 線上可呈現爆米花樣鈣化[20]。MRI 在T1WI 上表現為均勻的低信號,而在T2WI 加權像上表現為混雜信號或高信號,在T1 和T2 加權序列上硬化緣表現為低信號帶[21]。CT 主要表現為骨性膨脹伴特征性毛玻璃樣改變,有助于更好地顯示病變范圍,在評價基質礦化、假分隔、擴張、皮質變薄和病理性骨折方面更敏感[22]。ECT 表現為病灶處代謝異常活躍[23]。本病例X 線顯示左側髂骨、左側股骨上段髓腔內溶骨性病變,密度不均勻,見斑片狀低密度影及稍高密度影,呈皂泡樣改變,邊緣硬化不明顯,無骨膜反應。在MRI 上,病變表現為T1WI 低信號混雜點狀高信號,T2WI 及壓脂序列混雜高信號,DWI 高信號,增強掃描可見不均勻強化。組織學上,正常骨小梁及骨髓被大量增生的纖維樣組織所替代,其內散在分布著大小不等、形態不一的骨小梁,排列紊亂、無序,缺乏彼此連接,表面未見明顯骨母細胞襯覆。
Mazabraud 綜合征中的IM 臨床上可無任何癥狀,通常患者因腫塊不斷長大而就醫,偶爾可伴有疼痛。影像學上,IM 往往發生在FD 區域附近,通常表現為軟組織內界限清楚、均質的低密度團塊影。X 線呈非特異性軟組織腫塊,但由于敏感度較低,約一半病例可顯示正常結果[24]。MRI 表現包括T1WI上的低信號,T2WI 上的高信號,以及對比增強期的不同模式,包括不均勻或斑片狀和周邊增強等[25]。CT 通常顯示為邊界清楚的均勻軟組織腫塊,其密度高于水,低于周圍肌肉組織,大約50%的病例可見輕度彌漫性增強或周邊和間隔增強[26]。IM 的這些影像學特征與囊腫或其他黏液樣軟組織的良性/惡性腫瘤高度相似,鑒別難度較大,但如果存在多個病灶,應該考慮到一種綜合征的可能,如Mazabraud 綜合征或神經纖維瘤病。我們這例IM 為單發病灶,MRI 表現與文獻報道相符,T1WI 為低信號,T2WI 為高信號,增強掃描為環形強化,結合多發性骨損害,影像學上考慮為軟組織惡性病變。組織學上,腫瘤組織細胞稀少,呈梭形或星芒狀,無異型性,無核分裂像,間質血管稀疏,富含大量黏液樣基質;腫瘤與周圍組織分界不清,形成不完整的纖維包膜。免疫表型呈vimentin 陽性,CD34、SMA 和Desmin 不同程度表達,S100 陰性[13]。本例除vimentin 陽性外,其余標記均為陰性。IM 屬于良性間葉源性腫瘤,基本不惡變,與其他富含黏液、更具侵襲性的病變(如血管黏液瘤、黏液性纖維肉瘤、黏液性脂肪肉瘤等)的鑒別依賴于病變內缺乏高細胞密度、核分裂像和血管。對于形態學及免疫表型不典型的病例,GNAS1基因突變分析將有助于IM的精確診斷[4,27-28]。另外,在熟知Mazabraud 綜合征的情況下,相關的FD 的存在也提示軟組織病變為良性性質的可能。
Mazabraud 綜合征偶可同時伴發McCune-Albright 綜合征,后者臨床表現為多骨性FD、皮膚咖啡牛奶色素斑和內分泌功能異常[29]。McCune-Albright綜合征中的FD 均為多骨性病變,病因也與GNAS1基因突變有關[30]。皮膚色素斑一般分布在有骨病變的同側。內分泌功能異常可表現為性早熟、甲狀腺功能亢進、甲狀旁腺功能亢進、生長激素分泌過多、糖尿病、庫欣綜合征、多囊卵巢和乳腺纖維腺瘤病等。McCune-Albright 綜合征發病性別差異與Mazabraud 綜合征相似,也好發于女性,女∶男約為2∶1。但根據Biazzo 等[31]的病例報道和文獻復習以及后繼其他學者的個案報道[32-33],Mazabraud 綜合征同時伴發McCune-Albright 綜合征的患者幾乎均見于女性,13 例中僅1 例為男性,女∶男 > 10∶1,提示兩種綜合征伴同的患者比任一綜合征單發的患者,更常見于女性,但這種性別差異有待更多病例積累證實。
Mazabraud 綜合征的治療方面,對于IM 可行局部手術完整切除,在充分認識這一綜合征的情況下,可避免因誤診為惡性而導致的過度擴大切除。FD若為無癥狀病變,一般不需要治療,只需連續隨訪。癥狀性FD 的治療通常是非手術治療,包括雙膦酸鹽和非甾體抗炎藥等用于緩解疼痛[34]。 對于高應力部位(如股骨近端)有癥狀的、較大的或進行性的病變,以及非手術治療無效的患者,多主張手術治療。 FD 手術的目的是預防或治療畸形和穩定病理性骨折[35]。最常用的手術方法為刮除和植骨術,加或不加內固定[36-37]。但由于手術常難以取得滿意效果,目前還沒有有效的、成熟的醫學治療方法。最近,有研究顯示地舒單抗(denosumab)可直接靶向作用于FD 病變中異位的破骨細胞,促進基質細胞向成骨細胞分化,從而抑制病變發展[38],同時能夠有效緩解雙膦酸鹽治療無效的FD 患者的疼痛[39],因而denosumab 被認為是治療FD 前景看好的藥物。但停藥后導致的反彈相關骨折、頜骨壞死及危及生命的高鈣血癥等副作用,使denosumab 的臨床應用受到限制,后續需要進一步的研究來確定治療的最佳劑量和持續時間[40]。FD 因經放療后易誘發惡變,因而禁忌放療。
Mazabraud 綜合征的IM 一般不惡變,但在手術切除后可復發,通常認為其復發率低。但Majoor等[10]對Mazabraud 綜合征的20 例IM 患者在手術切除后進行了長期隨訪,發現其中6 例(30%)復發,復發時間為手術后1.9~16.0 年,中位年限為8.5年。這些研究結果提示Mazabraud 綜合征的IM 具有較高的復發率,而且術后復發似乎是一個非常緩慢而持久的過程。孤立性IM 和Mazabraud 綜合征的IM 均存在GNAS1 基因突變,提示兩者可能具有相同的發病機制。然而,Silver 等[41]對17 例孤立性IM 經手術切除后的患者進行平均7 年(1 至20 年不等)的隨訪,發現無1 例復發;而Mazabraud 綜合征的IM 如前所述,具有相對較高的復發率。這表明兩個實體之間存在差異。Mazabraud 綜合征中IM的較高復發率可能與組織學特征(如高細胞性)以及多發性IM 以不同速度發展有關,但確切因素仍有待進一步研究。
Mazabraud 綜合征的FD 為良性病變,偶可發生惡性轉化,形成骨肉瘤、軟骨肉瘤或高級別梭形細胞肉瘤[42],但依據報道的整體數據,目前認為其惡變率較高,可達6%[43-44]。因此,對Mazabraud 綜合征患者進行影像學長期監測是十分必要的,其中全身MRI 是目前首肯的最佳監測方法。本例經每半年一次的MRI 復查監測,至今隨訪1 年10 個月,未見IM 復發或FD 惡性轉化。
總之,Mazabraud 綜合征雖罕見,但影像學表現具有一定特征,最終確診依賴于病理學檢查。與孤立性FD 多發生在兒童和青少年期不同,Mazabraud綜合征中的FD 多出現在較晚的成年期。因此,對于發病年齡上可疑的FD 患者,應注意有無合并IM。反之,對于IM 的患者,也須警惕有無FD 存在。遵循從“懷疑”再到“驗證”這一診斷思維模式,Mazabraud 綜合征診斷的準確性和檢出率將會得到進一步提高,從而有效避免誤診、漏診。
作者聲明:本文全部作者對于研究和撰寫的論文出現的不端行為承擔相應責任;并承諾論文中涉及的原始圖片、數據資料等已按照有關規定保存,可接受核查。
學術不端:本文在初審、返修及出版前均通過中國知網(CNKI)科技期刊學術不端文獻檢測系統的學術不端檢測。
同行評議:經同行專家雙盲外審,達到刊發要求。
利益沖突:所有作者均聲明不存在利益沖突。
文章版權:本文出版前已與全體作者簽署了論文授權書等協議。
猜你喜歡
英語世界(2023年6期)2023-06-30 06:29:10
鴨綠江(2021年35期)2021-04-19 12:24:18
中國生殖健康(2020年2期)2021-01-18 02:51:26
考試與評價·高一版(2020年6期)2020-11-02 02:45:24
中國生殖健康(2019年3期)2019-02-01 06:12:26
小學生導刊(2018年13期)2018-06-29 03:49:00
現代檢驗醫學雜志(2016年4期)2016-11-15 02:01:14
鑿巖機械氣動工具(2016年3期)2016-03-01 04:00:25
海軍航空大學學報(2015年3期)2015-11-11 17:20:00
中國神經精神疾病雜志(2014年1期)2014-03-01 03:23:22
