過渡金屬催化氯代物的羰基化反應研究進展
2023-10-14 07:52:04王鵬史會兵趙德明馮保林陳倩楊妲
化工進展 2023年9期
關鍵詞:催化劑
王鵬,史會兵,趙德明,馮保林,陳倩,楊妲
(1 山東京博石油化工有限公司, 山東 濱州 256500;2 中國石油大學(華東)理學院,山東 青島 266580)
羰基化反應能夠在底物分子內高效構建羰基官能團而被廣泛關注,通過簡單的底物(烯烴、炔烴、烷烴、鹵代烴等基礎化學品)可制備多種高附加值化學品[1-9]。其中有機鹵代物作為底物也可制備結構多樣性的大宗化學品、精細化學品以及關鍵醫藥中間體[10-11]。有機鹵化物發生羰基化反應的活性順序不同,具體為C—F?C—Cl<C—Br<C—I活性依次增加(碳鹵鍵能影響),可以看出C—Cl鍵的活性相對較低而使其在溫和條件下難以高效高選擇性轉化[12],但是氯代物相對于溴代物和碘代物價格低廉,因此如何研發或者改善現有催化劑體系,實現氯代物溫和條件下的高效轉化或者拓寬更多類型氯代物的羰基化反應成為科研熱點[13-14]。本文綜述近年來不同類型氯代物與一氧化碳(CO)、各種親核試劑發生羰基化反應合成高附加值化學品的研究進展,同時對氯代物羰基化反應依然存在的挑戰和未來應用前景進行了展望(圖1)。
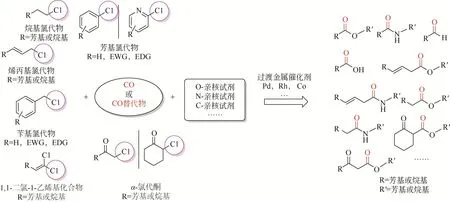
圖1 有機氯代物的羰基化反應轉化
1 氯代物羰基化反應機理
在CO 作為羰源的條件下,芳基鹵化物的氧化加成機理不同于鈀催化偶聯過程中相同芳基鹵代物的氧化加成機理[1,15-16]。這種差異是由無CO 配位的活性Pd(0)絡合物和有CO 參與配位的非活性Pd(0)羰基加合物(或簇)之間的含量不平衡造成的[1]。雖然雙齒膦配體可以有效阻止Pd(0)化合物的聚集,但在CO 存在的條件下,膦配體以及CO 分子(一個或兩個)依然可以和Pd(0)配位形成包含橋接CO 部分的雙金屬二聚體,從而影響反應活性[1,17]。
以鈀催化劑為例,氯代物發生羰基化反應的機理如圖2。首先是CO、配體氛圍中鈀絡合物的形成,其次經歷氯代物與鈀催化劑的氧化加成,再次CO 配位及插入形成酰基鈀活性中間體,在這一步,CO 遷移插入方形平面芳基氯化鈀配合物的速度很快,并且已被提出通過五配位中間體發生[1,18]。最后發生親核試劑進攻,鈀催化劑還原消除,同時反應體系中的堿締合氯化氫而形成目標產物。
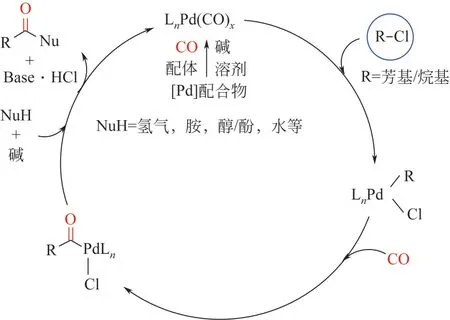
圖2 鈀催化芳基氯代物與親核試劑羰基化反應機理
在CO存在下,氯代物與Pd(0)氧化加成的機理研究報道很少。但是由于氯化物相比其他鹵化物便宜并且更具商業性,因此其羰基化反應也是研究熱點[19-20]。研究發現,芳基氯化物參與Pd(0)氧化加成的步驟以及羰基化反應本身的速率都很慢,這是因為芳基氯化物與低配位、富含電子的Pd(0)物種發生氧化加成反應速率最快,而CO 的結合使電子云密度較低,配位數增高,因此也抑制了反應速率[1,20-22]。由于芳基或雜芳基氯化物的羰基化需要較高溫度(>100℃),這也可能會導致CO 從Pd(0)配合物中解離或Pd(0)羰基簇的解離[1,23-26]。因此,更好地了解與芳基氯代物反應的金屬配合物的特性,可以發現新的催化劑體系,從而誘導該類底物的溫和羰基化轉化。
2 芳基氯代物的羰基化反應
芳基氯代物的羰基化轉化由于其價格廉價且產物用途廣泛獲得而備受關注[27-37],然而C(sp2)—Cl鍵較高的離解能導致其相對于溴代物和碘代物更難活化(室溫條件下,PhCl、PhBr和PhI的C(sp2)—X鍵能分別為402kJ/mol、339kJ/mol和272kJ/mol)[28-32]。
2.1 甲酰化反應
早在1982 年,Pétrier 等[33]報道了溫和條件下N,N-二甲基甲酰胺(DMF)作為羰源的芳基氯(氯苯)和烷基氯(1-氯丁烷、環己基氯)的甲酰化反應合成醛類化合物。經過條件篩選,當四氫呋喃(THF)作為溶劑時,目標產物的收率為70%~78%。2021年,Xiao等[34]也報道了一例DMF作為羰源(同時作為溶劑),CuI 催化氯苯可實現甲酰化高效合成苯甲醛(收率86%),同時該催化劑體系對烷基/芳基溴代物具有中等到優異的甲酰化底物普適性(45%~96%)。
2015 年,Iranpoor 等[35]首次使用五羰基鐵作為羰源實現了鈀催化芳基鹵化物的甲酰化反應合成芳香醛和氘代醛。該反應在常壓條件下以DMF 的水溶液作為溶劑,不需要添加氣態氫或任何還原劑便可生成醛類化合物。有趣的是芳基碘代物可在無膦配體的情況下發生反應[式(1)]。
另一方面,也有CO 作為羰源的研究報道,例如1989 年,Huser 等[36]報道了鈀催化芳基氯代物的甲酰化反應。當氯苯作為底物時,經過條件篩選(trans-[Pd(PC6H11)3)2(C6H5)Cl]/三環己基膦作為催化劑,添加三乙胺(Et3N)作為堿,甲苯作為溶劑,1.5MPa 合成氣),苯甲醛的收率高達90%。同年,Ben-David 等[37]也報道了一類富電子并且螯合穩定的鈀催化劑(dippp)2Pd(0)催化氯苯的甲酰化反應,該體系選擇甲酸鈉替代氫氣可實現苯甲醛95%的高效合成。
2.2 羰化胺化反應
在已知制備芳香酰胺的各種方法中,芳基碘化物和溴化物與CO、N-親核試劑的羰化胺化反應報道較多[38-45]。而使用芳基氯化物作為起始原料的報道相對較少[27,46-47],該領域的大部分工作均使用缺電子(或N-雜)氯代芳烴作為起始原料[40,48-54]。目前為止,只有兩種方案(一步法策略和串聯策略)應用于氯苯或含有給電子基團的芳基氯化物的羰化胺化反應[15,55-58]。
對于一步法策略,文獻報道中大多選用氨氣(NH3)作為親核試劑,而其他類型的伯、仲脂肪族和芳香族胺作為親核試劑的報道極少[15,55]。2010年,Wu 等[55]報道了鈀催化芳基和雜芳基鹵化物(22 個實例)與CO、氨氣(NH3)作為胺源的羰化胺化反應。該催化劑體系同樣適用于非活化的芳基氯化物(8 個實例),在溫和條件下以30%~60%的收率生成所需目標酰胺[條件1,式(2)]。2018 年,Wang 等[15]探究了乙烯基鈀絡合物(DCPP)Pd(C2H4)作為催化劑催化芳基氯化物與氨氣的羰化胺化反應的機理。該工作中,氯苯以83%的產率轉化為苯甲酰胺,缺電子/富電子的芳基氯化物均可以64%~100%的產率生成相應的苯甲酰胺[條件2,式(2)]。
對于串聯策略,2007年,Martinelli等[56]開發了一種通用的、耐官能團的、溫和的體系,用于鈀催化芳基氯化物串聯合成酰胺的羰基化反應。該反應在低CO壓力(0.1MPa)和中等溫度(110℃)下進行,通過酚鈉的引入,一方面促進了苯甲酸酯作為反應中間體的形成,另一方面可以作為隨后與胺反應的離去基團[式(3)]。該體系所選用的膦配體(DCCP·2HBF4)廉價、在空氣中穩定且商業可得,具有潛在的工業化放大前景;另一方面,他們通過原位紅外光譜詳細探究了酚鈉在反應過程中的作用。
2018 年,Lagueux-Tremblay 等[57]利用串聯的策略報道了鈀催化芳基氯化物和4-二甲氨基吡啶(DMAP)的羰化胺化反應。該反應首先生成芳酰基DMAP 鹽作為強親電體,其次伯胺和仲胺的親核進攻生成具有良好官能團相容性的酰胺[式(4)]。與通常需要親核試劑與弱親電鈀酰基中間體反應的經典羰基化反應不同,芳酰基DMAP 鹽的高親電性允許多種底物的酰化。這種轉化是由“Pd/Xantphos”催化劑介導的,機理研究表明,膦配體位的空間位阻效應影響其與Pd(0)的結合,從而影響ArCO-DMAP 產物的還原消除和高鍵能芳基碳氯鍵的氧化加成過程。總之,這種轉化允許芳基氯化物與一系列親核試劑以良好的官能團相容性生成酰胺和酯。
2022 年,Wang 等[59]首次報道了鈀催化低活性芳基氯化物(包括富電子、中性和缺電子的芳基氯代物)與伯、仲脂肪族和芳香族胺的直接羰化胺化反應。 該轉化成功的關鍵是在“Pd(OAc)2/Xantphos”催化劑體系中引入氯化銫(CsCl)作為添加劑。該催化體系實現了多種芳基氯化物與胺的有效轉化合成酰胺,并具有良好的官能團相容性(60 個實例,轉化率高達99%,分離收率高達95%)[式(5)],同時也有效改善了該類型的底物需要通過串聯策略合成目標產物的弊端。研究表明,親核試劑胺的用量會影響一氧化碳遷移形成酰基鈀中間體的過程,過量的胺會抑制羰基化產物的生成;并且該反應體系中一氧化碳的壓力(0.2MPa)過高或者過低均不利于反應的發生。
2.3 羰化酯化反應
醇通常比胺酸性更強,因此在堿性條件下,過渡金屬催化芳基氯代物可以原位生成醇鹽,并且醇或醇鹽可作為偶聯試劑繼續發生后續反應。相反,胺作為親核試劑很難形成陰離子酰胺鹽。另一方面,醇和醇鹽的配位能力和親核性與胺的也不相同,這便導致酰基配合物與氧基親核試劑的反應機理與氮基親核試劑的反應機理有所不同,發生羰基化反應的難易程度也有所差異[15,58]。
早在1989 年,Ben-David 等[58]報道了鈀催化芳基氯代物的羰化酯化反應。該工作選擇 (dippp)2Pd作 為 催 化 劑 [dippp=bis(diisopropylphosphino)propane],可以高效高選擇性合成芳基羧酸、酯和酰胺類化合物。隨后1990 年,Dufaud 等[60]也報道了鈀催化芳基氯代物(氯苯和缺電子取代的氯苯)的羰化酯化反應制備多種酯類化合物,Pd/C 作為催化劑,但是反應條件苛刻,200℃反應50h 才能實現底物的轉化,因此發展較溫和條件下芳基氯代物的羰化酯化反應顯得尤為重要。
2001 年,Mágerlein 等[52]報道了氯苯類化合物的高效羰化酯化反應。該工作發現了一種新的鈀催化劑體系以良好到優異的收率催化缺電子、中性和富電子的芳基氯化物生成目標產物。所使用的配體商業可得,并且在空氣中穩定(條件1,圖3)。次年,M?gerlein 等[61]優化反應條件,發現“PdCl2(PhCN)2-PCy3”組成的催化劑體系亦能很好的催化芳基氯代物的羰化酯化反應(條件2,圖3)。所選用的膦配體“三環己基膦”更加廉價易得,且目標產物收率優異(68%~91%)。

圖3 芳基氯化物羰化酯化反應的高效催化劑
2010 年,Schareina 等[62]選擇一氧化碳替代物,實現了鈀催化芳基氯代物的羰化酯化反應。經過條件篩選:1,8-二氮雜雙環[5.4.0]十一碳-7-烯(DBU)作為堿,乙腈作為溶劑時,目標產物酯收率高達81%。在對CO 替代物篩選的過程中發現:甲酸乙酯作為羰源時,3,4-二甲基苯甲酸丁酯收率為56%,而甲酸異丙酯作為羰源時,由于空間位阻效應目標產物收率較低(34%)[式(6)]。另一方面,該策略將底物普適性拓展至含有富電子基團的芳基氯代物。
2017 年,Shimomaki 等[63]以CO2作為羰源,在可見光的驅動下,實現了鈀催化芳基鹵化物的羰化酯化反應。該催化劑體系可以避免使用當量的金屬還原劑,且底物普適性優異;對于芳基溴代物亦可以高效轉化,首次實現了大位阻的2,4,6-三異丙基溴苯的羰化酯化反應[式(7)]。
2020年,Ai等[64]報道了一種新型鈀催化芳基氯化物的羰化酯化方法以良好至優異的產率合成多種苯甲酸甲酯。該反應能夠發生主要有以下因素:
①使用LiOMe可以作為促進羰基化轉化的堿或共親核試劑;②使用Pd/C 作為催化劑,可以防止傳統的二價鈀前體被甲醇還原為零價鈀而團聚;③CO濃度不能過高,否則將直接導致反應終止[式(8)]。該策略的優勢在于通過一種催化劑體系實現了各種類型的芳基氯代物(包括富電子、中性和缺電子)羰化酯化反應一步法合成酯。
3 烯丙基氯代物的羰基化反應
烯丙基氯代物與CO 羰基化反應轉化為β,γ-不飽和酯或酰胺的報道較少[65-73]。早在1963年,Tsuji等[66]報道了化學計量的π-烯丙基氯復合物可以調控烯丙基氯在乙醇溶劑中與CO 結合發生羰基化反應生成丁烯酸酯。很快他們[67-73]發現催化量的氯化鈀也可以使烯丙基氯、烯丙基醇、烯丙基醚等作為底物促進該反應[式(9)]。
1964 年,Dent 等[74]報道了π-烯丙基鈀配合物催化烯丙基氯化物的羰基化反應。該反應收率優異(94%)[式(10)]。隨后1969年,Medema等[75]報道了這個反應的詳細動力學研究。但是該反應需要在較高溫度(130℃)下發生,同時催化劑用量較大,很難實現工業化應用。
1995 年, Yamamoto[76]報道了甲酸鉀參與鈀催化肉桂酰氯的羰基化反應合成β,γ-不飽和羧酸。反應通過18-冠-6活化甲酸鉀(HCOOK),反應溫度50℃較溫和,但是CO壓力5.0MPa偏高[圖4(a)]。同時,在低溫條件下使用PdCl2(PPh3)2可以實現2-取代烯丙基氯化物的雙羰基化過程[室溫條件二乙胺作為親核試劑,圖4(b)]。

圖4 鈀配合物催化烯丙基氯化物的羰基化和雙羰基化反應
2014 年,Zhang 等[77]首次報道了銥催化烯丙基氯化物的羰化胺化反應高效高區域選擇性合成氨基甲酸酯(支鏈產物與直鏈產物的摩爾比高達98/2)。該反應羰源為二氧化碳(0.1MPa)且反應條件溫和(三乙烯二胺DABCO 作為堿,甲苯作為溶劑,反應溫度15℃)[式(11)],但是產物的區域選擇性調控依然是難點。
2017 年,Wu 等[78]報道了溫和條件下,甲酸作為羰源,鈀催化烯丙基氯羰基化直接合成β,γ-不飽和羧酸的反應(60℃)。雖然這類底物比其他烯丙基衍生物具有更好的產率和線性選擇性,但底物范圍受到限制[式(12)]。
2022 年,Wang 等[79]報道了鈀催化烯丙基氯代物為底物的羰基化反應(羰酯化和羰化胺化)制備β,γ-不飽和酯/酰胺。該工作通過對關鍵反應參數(鈀催化劑前體、溶劑和堿等)進行篩選優化,可以在溫和條件下[Pd(OAc)2摩爾分數0.1%、CO 壓力0.2MPa、60℃]合成多種β,γ-不飽和酯/酰胺。該催化劑體系對烯丙基C—Cl鍵的活化表現出優異的化學和區域選擇性(圖5),產物中沒有支鏈的羰基化產物生產,且無需膦配體參與、催化劑用量較低,有望實現工業化應用;另一方面,該策略對于不同類型的親核試劑(醇、酚、脂肪胺、芳香胺)具有良好的兼容性,亦能實現復雜天然產物分子的改造合成。
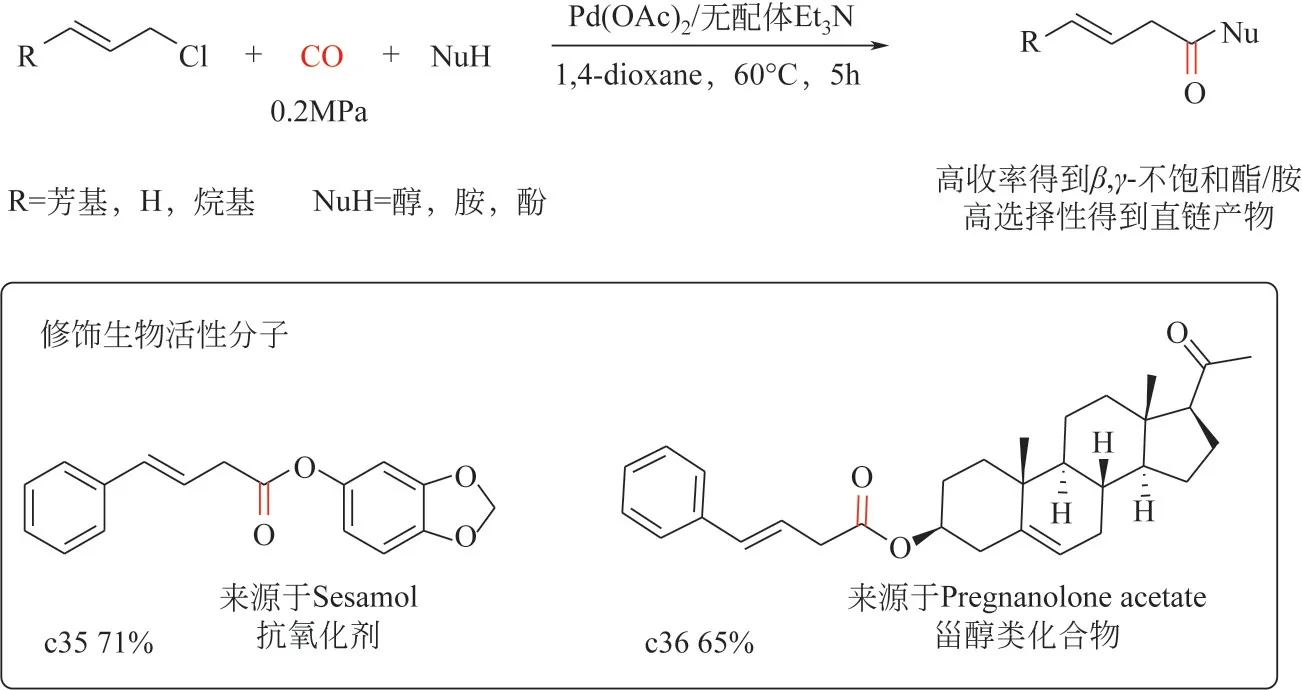
圖5 鈀催化烯丙基氯羰基化制備β,γ-不飽和酯/酰胺
4 芐基氯代物的羰基化反應
4.1 芐基氯代物羰基化反應
早在1974年,Schoenberg 等[80]報道了一類“乙酸鈀-三苯基膦”配合物在叔胺為堿的條件下催化氯化芐與CO、丁醇的羰化酯化反應。隨后,Hidai等[81]報道了鈀催化溴化芐的羰化酯化反應,鈀催化劑為Pd(CO)(PPh3)3、Pd3(CO)3(PPh3)4、Pd3(CO)3(PPh3)3和PdCl2(PPh3)2等。雖然該反應條件中需要引入一種以上的當量堿中和鹵化芐反應過程中產生的HX(X為鹵原子)。當然也有無堿反應體系的報道,例如,Urata 等[82]報道了分子篩類的沸石可以作為鹵化氫的有效吸收載體。
1983 年,Foà 等[83]報道了鈷催化芐基氯的羰化酯化反應,該催化劑體系65℃反應6h 收率高達65%。隨后1984 年,Tustin 等[84]報道了鐵催化芐基氯代物的羰化酯化反應。當甲醇作為親核試劑,碳酸鉀做為堿時,目標產物的收率高達65%。該反應在常壓室溫條件下反應過夜即可發生[式(13)]。
1989 年,Adapa 等[85]報道了溫和條件下芐基氯代物羰基化反應生成苯乙酸叔丁酯。作者對1-溴乙苯的羰基化反應也進行了研究[86],與氯化芐不同的是溴化芐對酯的選擇性較低。由于該類底物為仲碳取代的鹵代物,副產物除醚類化合物外,還可以在堿性條件下發生消除反應生成一定量的苯乙烯(10%~25%)。反應通過添加過量的配體,酯的收率可提高至70%[式(14)]。
除了合成酸和酯類化合物,鹵化芐可與其他親核試劑發生羰基化反應合成酮、酰胺等化合物。在磷酸鉀存在的情況下,Wu 等[87]利用商業可得的Pd(OAc)2/PCy3催化劑體系,報道了芐基氯化物與芳基硼酸的羰化偶聯反應生成α-芳基苯乙酮。值得注意的是,該反應以水作為溶劑[式(15)]。隨后,Wu 等[88]將親核試劑擴展到三氟芳基硼酸鉀,可以避免偶聯副產物的生成。
2011 年,Wu 等[89]成功地將芐基氯化物應用于鈀催化末端炔烴的羰化反應中。該反應中芳香族炔烴與脂肪族炔烴都具有很好的普適性。當芐基乙炔作為偶聯劑時,1,4-二芳基-3-丁-2-酮或取代呋喃酮的產量較好。該反應成功的關鍵是使用了缺電子的亞磷酸酯配體(圖6)。

圖6 芐基氯化物與末端炔烴的羰基化偶聯反應
2010 年,Troisi 等[90]發現在合成β-內酰胺的反應中目標產物收率較低,有大量相應的非環狀酰胺生成。他們認為是亞胺分解為胺類化合物導致的,胺進一步與酰基鈀中間體反應生成酰胺。基于這一發現,他們開發了一種鈀催化由芐基鹵代物和胺羰基化合成酰胺的新方法。反應形成酰基鹵化鈀中間體后,經歷來自胺的酰基親核取代反應而生成目標產物酰胺。當芐基氯代物作為底物時,酰胺的選擇性高達97%;烯丙基氯代物作為底物時,目標產物選擇性高達92%[式(16)]。
2012年,Wu等[91]報道了氨氣作為胺源的Pd催化氯化芐的羰化胺化反應。該反應通過對膦配體和催化劑前體的篩選,以中等至優異的收率(75%~98%)合成了不同類型的一級酰胺類化合物。在這個反應中,氨具有雙功能性:不僅用作胺化試劑,而且用作堿[式(17)]。
2020 年,Richardson 等[92]報道了鈀催化芐基氯代物的羰化胺化反應。該反應通過對溶劑、堿以及催化劑前體進行篩選,使用廉價的膦配體(DPEPhos)在較低溫度(70℃)下實現了底物與伯胺/仲胺的羰基化反應,該反應能夠抑制偶聯副反應的發生,高效高選擇性地合成2-芳基乙酰胺類化合物(圖7)。該催化劑體系中膦配體亦商業可得,且底物普適性廣泛,具有較好的官能團兼容性,為復雜的化學品的合成提供了新的方法。

圖7 鈀催化芐基氯代物的高效羰化胺化反應
在合成雜環化合物方面,基于鹵代烯丙基底物和亞胺的[2+2]環加成羰基化工作[93]:2009 年,Troisi 等[94]報道了鈀催化鹵化芐化合物羰基化和亞胺立體選擇性合成3,4-二芳基β-內酰胺;芐基溴的反應速度比芐基氯化物快,但β-內酰胺產率接近[圖8(a)];2013年,Wu等[95]報道了以水楊酸醛和芐基氯化物為原料,鈀催化羰基合成色甲酮的方法,以中等到優異的收率分離出多種香豆素(30%~95%)[圖8(b)]。
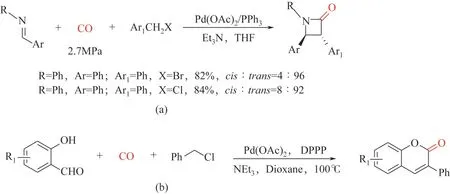
圖8 以鹵化芐為底物合成雜環化合物3,4-二芳基β-內酰胺以及色甲酮
4.2 氯化芐羰基化反應的工業化應用
芐基氯代物羰基化反應關注度最高的底物為氯化芐,其下游的苯乙酸以及苯乙酸酯類化合物在生活生產中具有重要的用途,例如可以制備重要醫藥青霉素工業鹽(PGK)如阿莫西林、頭孢氨芐、頭孢拉定、舒巴坦、他唑巴坦等抗生素的中間體[96-103]。工業生產中,苯乙酸的生產方法主要有苯乙腈水解法、苯-醋酐法、乙苯氧化法、苯-甲醛羰基化法、苯乙酰胺水解法等多種策略[96,98-100]。但是都存在以下弊端:反應過程中副產物較多,對操作環境污染嚴重,產品收率低,生產成本高。而芐氯羰基合成法(芐氯在氫氧化鈉和有機溶劑兩相體系中進行羰基化反應,生成苯乙酸鈉,然后在酸性條件下被酸化成苯乙酸)可高效制備苯乙酸而備受關注。該策略相較于傳統通過氯化芐和氰化物反應得到目標產物的方法具有如下的優點:①避免使用氰化鈉及中間體苯乙腈等劇毒物品,同時可以避免苯乙腈合成工序中產生易揮發劇毒且帶有惡臭的物質異氰芐,減少人員健康危害,減少這些物質對環境造成嚴重污染;②有效改善反應過程中的廢水污染;③改善苯乙腈合成過程中收率低的弊端[97,99-103]。但是該反應以鈀催化劑為主,由于催化劑價格昂貴,如何解決羰基化過程中面臨的成本問題便成為研究重點[98-101]。最主要的思路便是利用兩相體系實現原料、催化劑與產物分離,進而實現催化劑的多次循環利用。
Cassar 等[96]報道了鈀催化兩相體系中鹵化芐發生羰基化反應一步法生成相應的酸。該策略將氯化芐和PPh3緩慢加入氫氧化鈉(30%)的水溶液中,同時加入Pd(PPh3)2Cl2和n-Bu4I,在95℃、CO 壓力0.5MPa 條件下制備苯乙酸,產率高達83%。值得注意的是,該反應體系的催化劑可以多次循環使用而沒有明顯的流失(圖9),因此該策略有望實現原料、催化劑與產物的分離,并且大規模生產應用。
Okano 等[97]在Cassar 兩相催化劑體系的基礎上報道NaOH/庚烷兩相體系,使用磺化的膦配體將鈀絡合物溶解在水相中。該方法后續被Kohlpaintner等[98]進一步發展,使用Pd(OA)2(或PdCl2)以及磺化膦配體,催化氯化芐在溫和條件(CO 壓力0.1MPa)下產生相應的苯乙酸,產率為80%~94%。反應后水相經有機溶劑萃取便得到羧酸鈉鹽[轉化數(TON)>1500;轉化頻率(TOF)達到135h-1]。隨后經過酸化產生游離酸產物(圖10)。該策略很好地實現原料、催化劑與產物分離。

圖10 鈀催化芐基氯代物羰化酯化反應合成苯乙酸
1999 年,Pellegrini 等[99]除了使用CO 和甲 醇使氯化芐進行羰基化反應外,選擇CO 替代物甲酸甲酯作為羰源,Mg(OMe)2誘導甲酸甲酯脫碳促進反應的完成。他們發現添加額外的CO 會抑制羰基化過程[圖11(a)]。隨后2001年,Gavi?o等[100]通過優化反應條件再次報道了溫和條件下鈀催化芐基氯化物的羰化酯化反應,并且探究了CO 壓力對該反應的影響。他們在沒有CO 的情況下加熱反應介質(避免形成鈀羰基物種,高濃度的該物種不利于羰基化過程),利用鎂形成甲氧基離子,再通入CO,便可以在較低的CO 壓力(30℃、0.3MPa)和適當的攪拌速度下,避免催化劑失活從而促進反應的發生[圖11(b)]。2006年,Pryjomska 等[101]也報道了一例較溫和條件下(60℃、CO 壓力為0.5MPa)鈀催化芐基氯的羰化酯化反應,但是產物苯甲酸甲酯的收率較低(15%)[圖11(c)]。如何在溫和條件下提高目標產物的收率也是該策略的研究難點。

圖11 溫和條件下芐基氯的羰化酯化反應
除了鈀催化劑體系,鈷系催化劑亦能很好地催化芐氯羰基合成苯乙酸[102-106],該催化劑體系已經實現工業化應用。當Co2(CO)8作為催化劑前體,反應可在較溫和(30~70℃)、CO 壓力0.1~1.0MPa 條件下發生,苯乙酸收率可達85%~90%。當四羰基鈷的鈉鹽為催化劑前體時,反應在水油兩相中進行,生成苯乙酸鈉中間產物,經過酸化后得目標產物,收率一般為80%~90%。選擇適當的助劑可使苯乙酸鈉不斷地從水相中分離出來,催化劑完全溶于有機相中循環,從而實現生產過程的連續化。該方法的優點如下:①催化劑制備貯存及循環較容易,回收率高;②羰基化反應在低壓條件下即可發生;③苯乙酸收率較高,重結晶后產品純度高;④CO 利用率高,三廢較易處理(其中廢鹽水經處理可用于氯堿廠作電解原料)[103]。
總之,為解決過渡金屬催化劑分離和循環使用的方法(均相催化多相化)可歸納為兩大類:一類是將催化劑靜態固定在高分子或無機載體上,形成固載化均相催化;另一類則是采用功能化的膦配體(水溶性膦配體或離子化的膦配體),將均相催化劑動態鎖定在與產物互不相溶的水相(或者離子液體)中,而實現水/有機兩相催化[107-108]。而該方法也為從源頭上解決金屬催化劑回收及三廢后處理等問題提供一個有效的工具[108]。另一方面,陳金龍等[109]也報道了一種苯乙酸生產廢水的治理與資源回收利用方法,它是將苯乙酸以及生產后廢水通過苯乙烯-二乙烯基苯共聚大孔吸附樹脂,使廢水中苯乙酸等大部分有機物吸附在樹脂上;吸附出的水無色透明,可送隔膜法電解車間配制電解鹽水以回收利用其中的氯化鈉。
5 α-氯代酮的羰基化反應
[Pd]/P配體體系催化的α-溴代丙酮的羰基化反應(特別是羰化酯化)在1975 年就有報道[85,110], 但是α-氯代丙酮的羰基化反應報道較少[13,111-113]。1989年,Adapa 等[85]報道了溫和條件下芳基鹵代甲基衍生物的羰基化反應制備叔丁酯,該工作只有一個實例為α-氯代丙酮羰基化生成相應的酯(收率為50%)。1999 年,Cavinato 等[114]報道了NEt3作為堿,PdCl2(PPh3)2-PPh3催化α-氯代環己基酮羰基化反應合成β-酮酯類化合物。該催化劑體系需要較高的CO壓力(10.0MPa),并且收率伴隨CO壓力的增加而增多[式(18)]。
2002 年,Lapidus 等[115]報道了三丁胺存在下,鈀催化鹵代甲基酮(X=Cl,Br)的羰基化反應合成乙酰乙酸烷基酯和β-芳基-β-酮酯類化合物,產率為68%~86%。該反應條件為CO 壓力1.0MPa,110℃反應2h完成[式(19)]。該反應體系對于氯甲基酮具有良好的羰基化化學選擇性,當2-溴苯乙酮做底物時,可產生大量的苯乙酮作為副產物。通過延長反應時間,該反應可以在一個大氣壓條件下發生。他們對反應機理進行了探究:羰基化過程中存在堿(Bu3N)不僅可以中和形成的氫鹵酸,而且可以催化酰基絡合物的醇解;另一方面,Bu3N 的濃度對反應速率的影響表明酰基絡合物的醇解是速率限制步驟。
2012 年,Wahl 等[116]采 用Pd(acac)2/Xantphos 催化α-氯酮發生羰基化反應合成β-酮酯,目標產物收率高達98%,該催化劑體系底物普適性良好,適用于不同類型的醇類、一級和二級α-氯酮類底物[式(20)]。他們通過對反應條件的優化,以使用比文獻報道的低得多的催化劑負載量(摩爾分數0.1%)高產率生成β-酮基酯,通過高壓核磁、紅外表征以及理論計算探究了催化循環過程中的活性中間體,并驗證了膦配體(Xantphos)的選用有助于提高目標產物的收率,是相較于堿、一氧化碳壓力、溫度等因素中最重要的影響反應活性的因素。
2014 年,Perrone 等[117]報道了芳香族亞胺和α-氯酮的立體選擇性羰基化反應合成具有N-芳基或N-烷基取代的α-亞烷基β-氧代酰胺。該反應可以一鍋法高立體選擇性生成Z式異構體的目標產物酰胺(24%~80%)[式(21)]。該方法可應用于含有N-芳基或N-烷基取代基的多種C-芳基亞胺,整個過程是高度立體選擇性的,并且僅提供作為(Z)異構體的α-亞烷基β-氧代酰胺,還提出了一種涉及酰基-β-內酰胺中間體的機制假說。
羰化串聯反應能夠以氫甲酰化的產物醛為中間產物進一步合成高附加值化學品,與多步反應相比,一鍋法串聯反應更為綠色和經濟。α-氯酮的羰化串聯反應由于能夠合成更高附加值產品也引起了廣泛關注[118-120]。2012 年,Wahl 等[121]報道了鈀催化α-氯酮串聯羰基化/烯丙基化加成反應合成烯丙基酮酯類產物。在第一步中,α-氯酮羰基化形成β-酮酯作為親核中間體,然后在4,5-雙(二苯基膦)-9,9-二甲基氧雜蒽(Xantphos)配體的調控下,與烯丙基苯甲酸酯反應以良好的產率獲得目標烯丙基化β-酮酯[圖12(a)]。同年,Giboulot 等[122]報道了鈀催化α-氯酮串聯羰基化/脫羧烯丙基化反應高選擇性合成γ,δ-不飽和芳香酮。該反應中,α-氯代苯乙酮首先與烯丙醇發生羰基化反應,然后中間產物原位脫羧生成相應的單烯丙基酮。該反應底物普適性優異,底物范圍可擴大到取代的α-氯代苯乙酮以及各種烯丙醇[圖12(b)]。

圖12 鈀催化的α-氯酮參與的羰化串聯反應
2013年,Wahl 等[123]報道了一類以α-氯酮為底物,通過簡單高效的串聯羰基化/邁克爾加成反應制備α-烷基β-酮酯類產物(圖13)。他們通過條件優化可以在一鍋反應中使用相同的堿以良好的產率(高達86%)制備高度官能化的α-烷基化β-酮酯,包括12 種原始產物。該多米諾反應的范圍可以擴展到具有各種邁克爾受體的伯和仲氯酮,包括丙烯酸甲酯、甲基乙烯基酮和環烯酮等。

圖13 α-氯酮的羰基化/邁克爾加成串聯反應
6 1,1-二氯-1-乙烯基化合物的羰基化反應
文獻報道在堿存在的情況下,烷基四羰基鈷配合物RCo(CO)4可在溫和條件下催化芳基鹵化物的羰基化反應,從而高效合成烷基芳基酮、羧酸和α-氧代羧酸的混合物。該反應也被應用于鹵代乙烯的羰基化反應,得到相應的羧酸。早在1989年,Miura等[124]報道了氫氧化鈉作為堿,八羰基二鈷與碘甲烷反應原位生成的四羰基甲基鈷作為催化劑,催化1,1-二氯-1-乙烯基化合物進行羰基化反應,高效合成丙烯酸類化合物。當甲醇和水作為溶劑時,目標產物選擇性高達68%。
后來,α-鹵代丙烯酸酯被廣泛應用于有機合成和醫藥中間體的生產中,后續可進行多種轉化,例如發生邁克爾反應;環加成合成噁唑、吡唑或環谷氨酸鹽;α-鹵代丙烯酸酯的C—C雙鍵也可以對映選擇性氫化產生α-氯代、α,β-不飽和醛或烯丙醇[125-129],這些通過轉化得到的高附加值產品已用于制備藥物或用于天然產物合成中[130-133]。但是高效合成α-氯代丙烯酸酯的報道很少:Wittig 反應、Horner Wadsworth-Emmons 反應、氯甲硅烷基烯酮衍生物與醛反應制備等[134-137],通過羰基化反應策略制備的案例更少。
2010年,Arthuis等[138]以1,1-二氯-1-烯烴為底物,通過鈀催化的羰化酯化反應高效合成了一系列Z-α-氯代丙烯酸酯。反應條件溫和(CO 壓力常壓、70℃、24h),目標產物收率55%~91%[式(22)],該反應具有良好的官能團兼容性以及廣泛的底物普適性。但是對于1,1-二氯-1-烯烴為底物的羰化胺化反應、甲酰化反應均沒有報道。
7 烷基氯的羰基化反應
相比于芳基氯代物的羰基化反應,烷基氯代物的羰基化反應報道很少。經過文獻調研,烷基碘化物和溴化物的羰基化反應已有大量報道,而過渡金屬催化的烷基氯化物作為底物的羰基化反應仍然是此類轉化的挑戰[4,139]。
對于烷基氯代物的羰基化反應,早在1989年,Huser 等[140]就報道了二氯甲烷參與鈀催化羰基化反應的案例,同時也提到了羰基化反應過程中對氯苯的活化。但是烷基氯代物的氫甲酰化反應還沒有報道,目前只有烷基溴代物和碘代物的氫甲酰化反應報道[141-142]。2007年,Jia等[143]報道了KI存在下,鈷配合物[Co(OAc)2、CoCl2]催化氯代烷烴與CO的羰化酯化反應。結果表明,Co(OAc)2的催化活性高于CoCl2,堿性添加劑NaOAc 有利于反應的進行。有趣的是,使用NaOAc 作為添加劑,Co(OAc)2和CoCl2表現出類似的催化活性。機理研究表明碘離子的作用最初是通過原位取代氯代烷烴中的氯原子形成活性碘代烷烴,然后在光照作用下進行碘代烷基化合物的羰基化轉化[式(23)]。
同年,Wang 等[144]報道了銅和鎘鹽結合組成的光催化劑(CuBr2、CuCl2和CdI2)體系催化氯代烷烴與CO 的羰化酯化反應。在這些催化劑中,CdI2催化底物生成的目標酯產率和選擇性最高,特別是氯代環己烷羰基化的產率和選擇性分別達到60%和75%。此外,在CuBr2和CuCl2催化劑體系中加入三正丁胺可以提高酯的產率和選擇性。有趣的是單一的CdCl2不進行羰基化反應,但添加NaI·2H2O后,CdCl2的催化活性有所提高(圖14)。
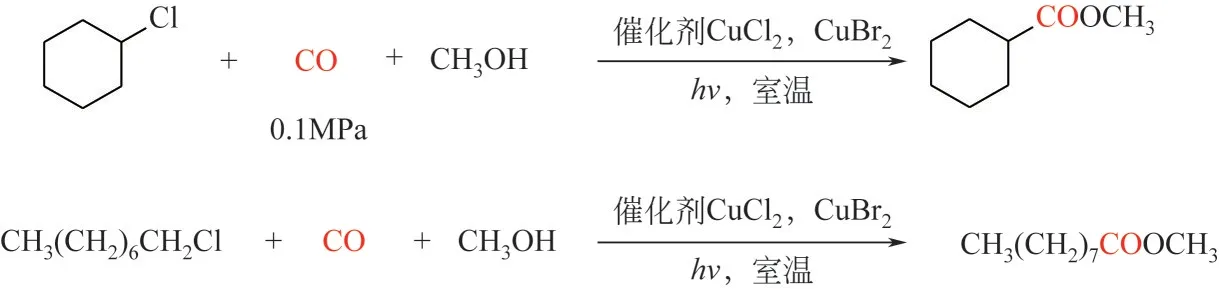
圖14 非貴金屬催化劑催化氯代烷烴與CO的光催化羰基化反應
2022 年,Wang 等[145]首次報道了銠催化未活化烷基氯化物的羰化酯化反應(脂肪族醇或酚均可作為親核試劑)制備酯類化合物。該反應之所以能發生是因為碘化鈉的添加使得氯代物原位轉化為活性更高的碘代物中間產物(Monsanto 工藝)。“Rh(acac)(CO)2/DPPP”催化劑體系可一鍋法直接制備多種脂類化合物(81 個實例,分離收率最高至95%);當一氯甲烷或者二氯甲烷作為底物時,該催化劑同樣可以高效合成多種乙酸酯類化合物。DPPP 的引入不僅可以調控羰基化反應的選擇性,還可以調控第一步鹵化物交換反應的選擇性(圖15)。

圖15 銠催化非活化烷基氯代物的羰化酯化反應
8 結語
氯代物的羰基化反應的產品在大宗化學品、精細化學品、日用品的合成以及藥物分子的設計合成中具有廣泛的應用。本文綜合評述了近年來不同類型的氯代物(芳基氯代物、烯丙基氯代物、芐基氯代物、α-氯代酮、1,1-二氯-1-乙烯基化合物、烷基氯代物)發生羰基化反應的研究進展。該類反應由于底物以及親核試劑的多樣性,可以合成諸多結構多樣性的醛、酯、羧酸、酰胺等在工業和藥物化學中有廣泛應用的化學品;同時合成路線簡單,可一步合成所需要的目標產物;目標產物的收率和選擇性高,一般均可以達到60%以上;氯代物相對其他鹵代物儲量豐富且廉價易得,產品具有高附加值,因此經濟效益可觀。
雖然目前氯代物的羰基化反應研究已經取得了很大進展,但該反應目前仍然存在許多問題亟待解決:①在CO存在下,芳基鹵化物與Pd(0)配合物的氧化加成機制尚不明確,酰基鈀配合物與親核試劑的反應機制也尚不清楚;②更深入地了解氯代物與金屬配合物的特性,發現更多新的羰基化反應和催化劑體系,誘導芳基氯代物或烷基氯代物的溫和羰基化;③均相催化中,氯苯以及烷基氯代物的氫甲酰化反應難度大,目前報道很少;④雖然光催化烷基氯代物的羰基化反應已有報道,但是如何通過加熱條件實現過渡金屬催化該反應的報道依然很少;⑤雖然一級烷基氯代物的羰基化反應已有報道,但是二級/三級烷基氯代物的羰基化反應(加熱條件)尚未有報道。⑥如何在實驗室提高目標產物化學/區域選擇性的基礎上,降低催化劑體系的成本,實現反應規模的放大乃至催化劑的多次循環依然需要探究。同時在工業化生產過程中,如何選擇合適的堿添加劑及時中和反應產生的氯化氫也是需要考慮的問題。總之,展望發展更多新型的催化劑體系實現更多類型氯代物的可循環羰基化高效和高選擇性轉化,實現更多下游高附加值羰基化學品的工業化應用。
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
