藏藥六味石榴丸的質量標準研究
2023-10-25 12:13:24周德來龍布才讓張明童
中國民族民間醫藥 2023年19期
李 運 周德來 龍布才讓 張明童
1.蘭州市食品藥品檢驗檢測研究院,甘肅 蘭州 730050;2.甘肅中醫藥大學藥學院,甘肅 蘭州 730000;3.甘南州合作市卡加曼藏藥開發有限公司,甘肅 合作 747000;4.甘肅省藥品檢驗研究院,甘肅 蘭州 730070
六味石榴丸源于《四部醫典》,藏文名“賽知周巴”,由石榴子100 g、肉桂50 g、肉豆蔻50 g、胡椒50 g、紅花50 g、大托葉云實50 g等六味藥材組成[1]。方中石榴子治一切胃病,能提升胃陽,并且可治療培根寒證;肉桂散熱止痛、溫通筋脈;肉豆蔻溫中行氣;胡椒溫中散寒;紅花活血通經、散瘀止痛;大托葉云實溫腎,逐寒等,諸藥共奏調節三因紊亂,溫腎、暖腰膝之功效。臨床常用于婦女白帶病。該方始源于人們實踐的直接經驗,有較牢靠的經驗基礎,在指導數千年醫療實踐活動中,得到不斷創新完善。為了提高藥效的持久性,減少吸潮,便于貯存,并增加服用及攜帶的便利性,以六味石榴散為基礎,將其劑型改變提升為丸劑(水丸),并建立較完善的質量標準,以保障藥品質量。課題組在前期工作的基礎上,結合相關文獻[2-9]研究,建立了處方中主要原料的顯微鑒別、薄層鑒別方法,并采用 HPLC 法分別對制劑中石榴子所含鞣花酸、紅花中羥基紅花黃色素A的含量進行測定,從而更加有效、系統地控制藏藥六味石榴丸的質量,確保臨床用藥安全、有效。
1 儀器與材料
1.1 儀器 BX53+DP73正置熒光生物顯微鏡(日本奧林巴斯株式會社);薄層色譜成像系統(瑞士CAMAG公司);Waters e2695高效液相色譜儀(美國Waters公司);XSE205DU型分析天平(瑞士Mettle Toledo公司);SB-800DT型超聲波清洗機(寧波新芝產品)。
1.2 材料 石榴子對照藥材(批號:121431-201002)、肉豆蔻對照藥材(批號:120926-201608)、紅花對照藥材(批號:120907-201713)、胡椒堿對照品(批號:110775-202107,純度:98.2%)、鞣花酸對照品(批號:111959-201903,純度:88.8%)、羥基紅花黃色素A對照品(批號111637-202111,純度:96.8%)均由中國食品藥品檢定研究院提供。六味石榴丸(批號:210801、210802、210803)由甘南州合作市卡加曼藏藥開發有限公司生產,陰性樣品由實驗室模擬制得;硅膠G、硅膠GF254、硅膠H薄層板(規格:10 cm×10 cm,默克公司);甲醇、乙腈(色譜純),水為Milli-Q超純水,其他試劑為分析純。
2 方法與結果
2.1 顯微鑒別 取六味石榴丸粉末適量,進行顯微制片(3~5張),置鏡下觀察可見:石細胞無色,橢圓形或類圓形,壁厚,孔溝細密(石榴子)。石細胞呈類圓形或類長方形,直徑約32~80 μm,一面壁菲薄(肉桂)[2,10]。石細胞淡黃色,成群或散在,呈類圓形或多角形,直徑20~35 μm,胞腔大,壁厚,木化,孔溝明顯(胡椒)。花粉粒圓球形或橢圓形,直徑約60 μm,外壁有刺,具三個萌發孔(紅花)。結果如圖1。

1.花粉粒(紅花);2.分泌細胞(紅花);3.石細胞(石榴子);4.石細胞(胡椒);5.石細胞(肉桂)
2.2 定性鑒別
2.2.1 石榴子的薄層色譜鑒別 取六味石榴丸粉末約1.0 g,置離心管中,加入乙醇溶液10 mL,超聲提取30 min,取其上清液作為供試品溶液;另取石榴子對照藥材1.0 g,按上述方法制備對照藥材溶液。依據處方配比及制備方法,完成缺石榴子陰性對照樣品的制備,照供試品溶液的制備方法,同法制成陰性對照溶液。吸取上述供試品及陰性對照溶液各5 μL,對照藥材溶液4 μL,點于同一硅膠GF254薄層板上,以石油醚(60~90 ℃)-乙酸乙酯(9∶1)作為薄層展開系統,展開,取出,晾干,置紫外燈(254 nm)下檢視。供試品色譜中,在與對照藥材色譜相應的位置上,應顯相同顏色的熒光猝滅斑點,且與陰性對照樣品中不存在干擾[11],如圖2所示。
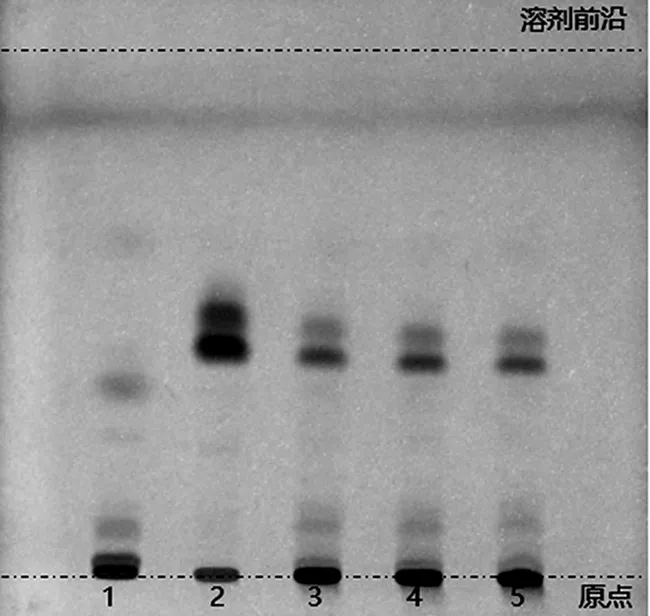
1.陰性對照樣品溶液;2.對照藥材溶液;3~5.供試品溶液
2.2.2 肉豆蔻的薄層色譜鑒別 取六味石榴丸粉末約1.0 g,置離心管中,加入石油醚(60~90 ℃)10 mL,超聲提取30 min,濾過,取續濾液作為供試品溶液。另取肉豆蔻對照藥材1.0 g,按上述方法制備對照藥材溶液。依據處方配比及制備方法,完成缺肉豆蔻陰性對照樣品的制備,照供試品溶液的制備方法,同法制成陰性樣品溶液。吸取上述溶液各5 μL,分別點于同一高效硅膠G預制薄層板上,以石油醚(60~90 ℃)-乙酸乙酯(9∶1)作為薄層展開系統,經預飽和后,展開,取出,晾干,噴以5%香草醛硫酸溶液,在105 ℃加熱至斑點顯色清晰。供試品色譜中,在與對照藥材色譜相應的位置上,應顯相同顏色的斑點,且陰性對照樣品中不存在干擾[11],如圖3所示。
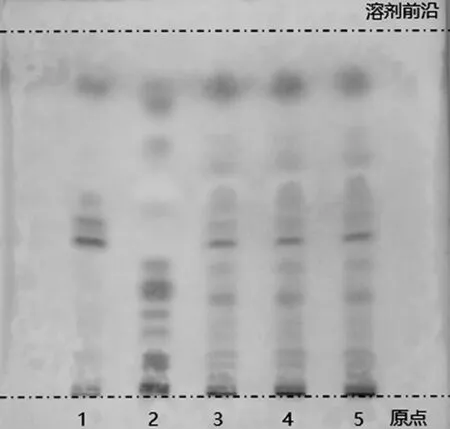
1.陰性對照樣品溶液;2.對照藥材溶液;3~5.供試品溶液
2.2.3 胡椒的薄層色譜鑒別 取六味石榴丸粉末約1.0 g,置離心管中,加入無水乙醇10 mL,超聲提取30 min,濾過,將續濾液作為供試品溶液。另取胡椒堿對照品,置棕色容量瓶中,加無水乙醇制成每1 mL含2 mg的溶液,作為對照品溶液。按處方比例及制備工藝,制備缺胡椒的陰性對照樣品,照供試品溶液的制備方法,同法制成陰性對照樣品溶液。吸取供試品及陰性對照樣品溶液各4 μL,對照品溶液2 μL,分別點于同一硅膠G薄層板上,以甲苯-乙酸乙酯-丙酮(7∶2∶1)作為薄層展開系統,展開,取出,晾干,噴以10%硫酸乙醇溶液,加熱至斑點顯色清晰,分別置日光和紫外光燈(365 nm)下檢視。供試品色譜中,在與對照品色譜相應的位置上,應顯相同顏色的斑點或熒光斑點,且陰性對照樣品中不存在干擾,如圖4所示。
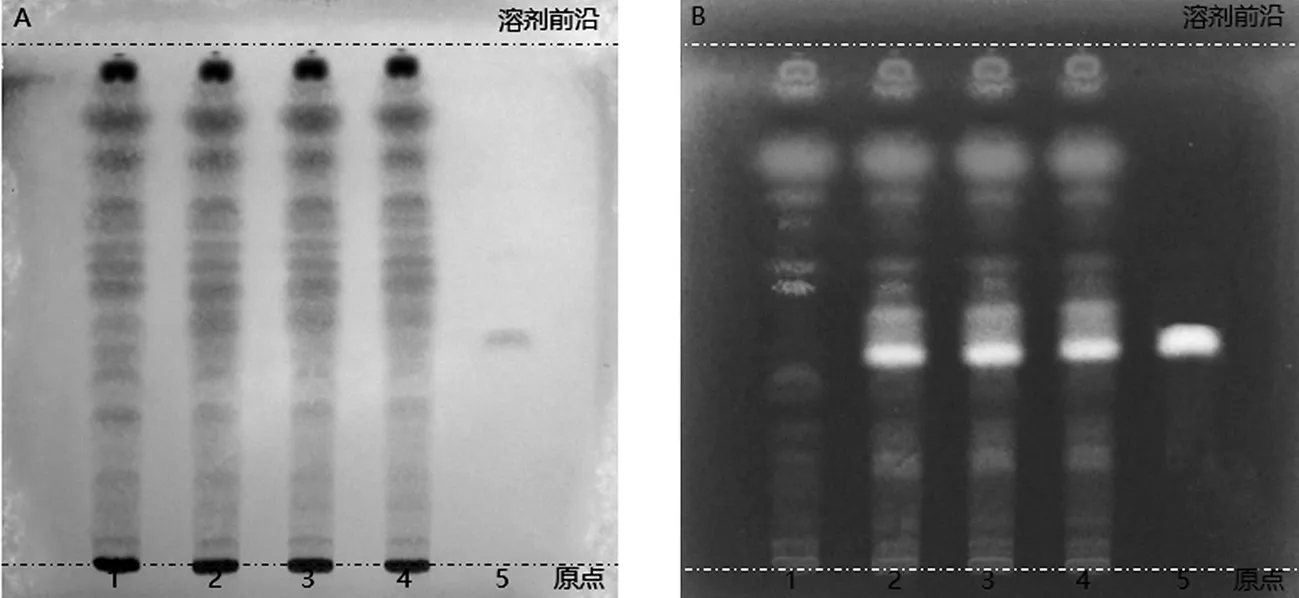
1.陰性對照樣品溶液;2~4.供試品溶液;5.對照品溶液;A.日光下檢視;B.紫外光燈(365 nm)下檢視
2.2.4 紅花的薄層色譜鑒別 取六味石榴丸粉末約1.0 g,置離心管中,加入80%丙酮溶液10 mL,密塞,振搖15 min,靜置后取上清液,作為供試品溶液。另取紅花對照藥材0.5 g,加入80%丙酮溶液5 mL,同法制成對照藥材溶液。依據處方配比及制備方法,完成缺紅花陰性對照樣品的制備,照上述供試品溶液的制備方法,同法制成陰性對照樣品溶液。吸取上述溶液各5 μL,分別點于同一硅膠H薄層板上,以乙酸乙酯-甲醇-甲酸-水(7∶0.4∶2∶3)作為薄層展開系統,展開,取出,晾干。供試品色譜中,在與對照藥材色譜相應的位置上,應顯相同顏色的斑點,且陰性對照樣品中不存在干擾,如圖5所示。
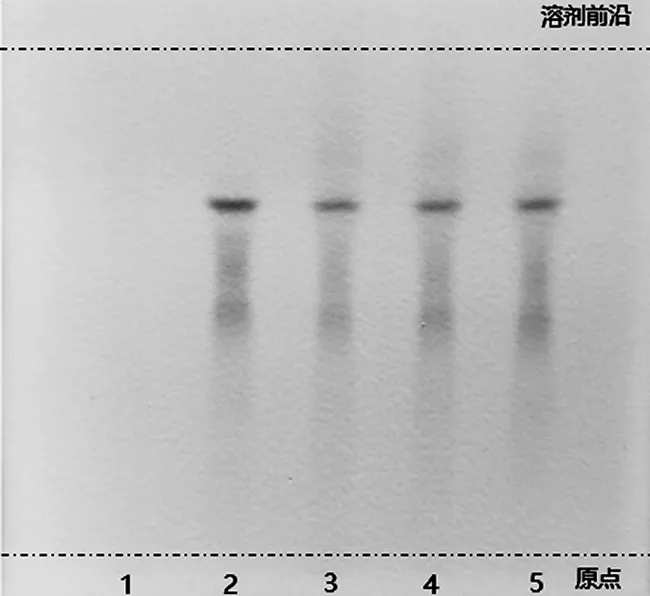
1.陰性對照樣品溶液;2.對照藥材溶液;3~5.供試品溶液
2.3 六味石榴丸中鞣花酸的HPLC含量測定
2.3.1 對照品溶液的配置 精密稱取鞣花酸對照品12.74 mg,置于25 mL容量瓶,用甲醇溶解、定容,搖勻。另取1 mL至10 mL容量瓶中,用甲醇定容至刻線即得對照品儲備液,備用。
2.3.2 供試品溶液的制備 取六味石榴丸適量研細,另取約1.0 g,精密稱定,置具塞錐形瓶中,精密加入甲醇50 mL,稱定重量,超聲提取(功率 300 W,頻率40 kHz)50 min,放冷,再稱定重量,用甲醇補足減失的重量,搖勻,靜置后,取上清液經0.22 μm微孔濾膜濾過,取續濾液作為供試品溶液。
2.3.3 陰性對照樣品溶液的制備 依據處方配比及制備方法完成不含石榴子藥材的陰性對照樣品的制備,按照“2.3.2”項下方法制備陰性對照樣品溶液。
2.3.4 色譜條件 CAPCELL PAK-C18(250 mm×4.6 mm,5 μm);柱溫:30 ℃;以乙腈-0.7%磷酸溶液(16∶84)為流動相;流速:1.0 mL/min;檢測波長:252 nm。理論板數按鞣花酸峰計算應不低于4000。
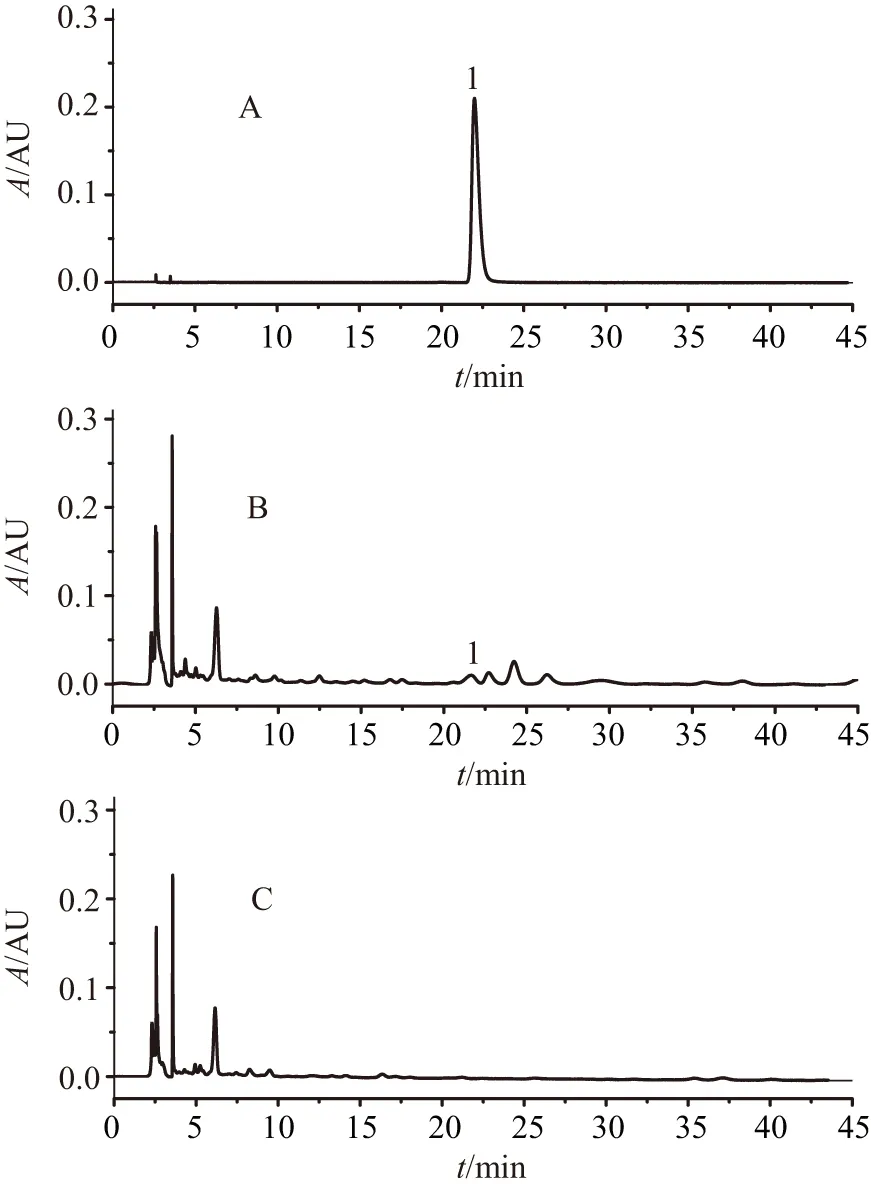
A.對照品溶液;B.供試品溶液;C.陰性對照樣品溶液;1.鞣花酸
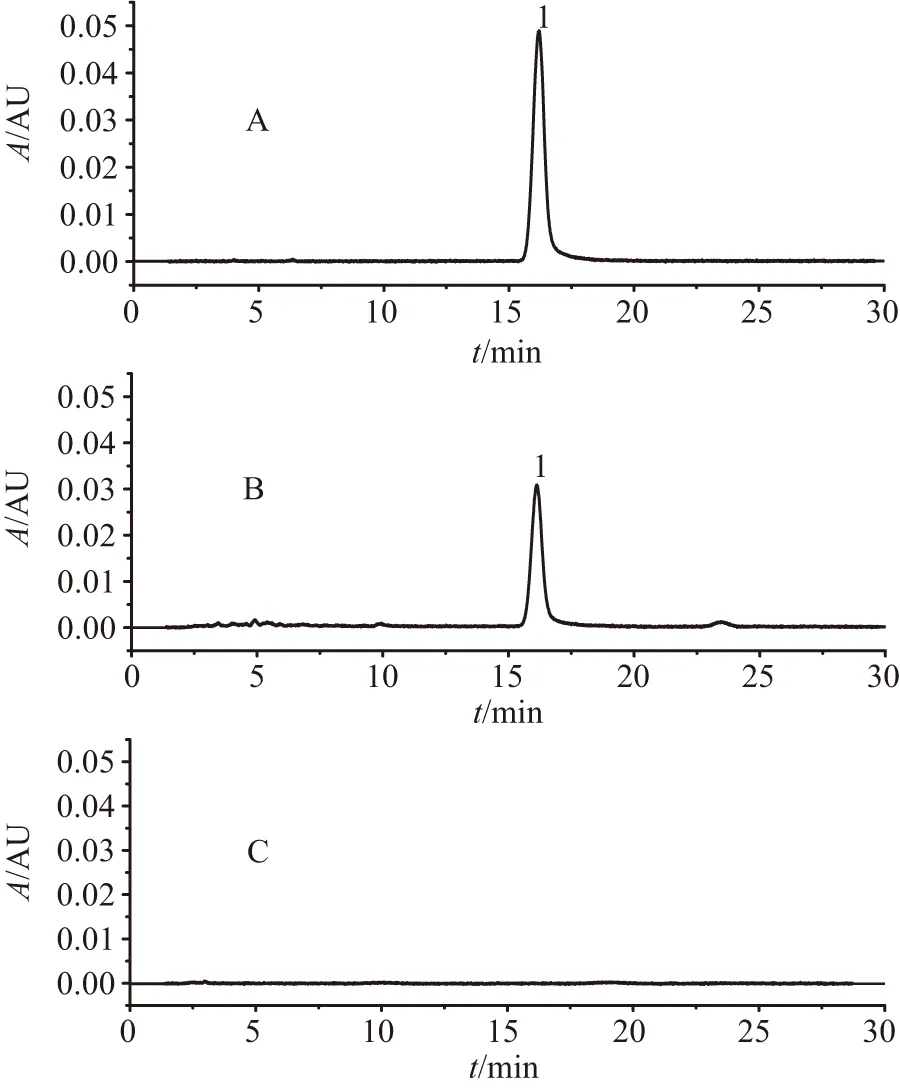
A.對照品溶液;B.供試品溶液;C.陰性樣品溶液;1.羥基紅花黃色素A
2.3.5 專屬性試驗 取上述對照品溶液10 μL、供試品溶液和陰性對照樣品溶液,各20 μL,按照“2.3.4”項下條件檢測。由圖6可見,目標化合物分離度良好;陰性樣品色譜圖中,鞣花酸對應目標化合物在相應的保留時間無色譜峰吸收出現,表明處方中其他共存成分對含量測定結果不存在相互干擾。
2.3.6 線性關系考察[12]分別精密移取“2.3.1”項下備用儲備液0.125 mL、0.25 mL、0.5 mL、1.0 mL、2.0 mL置于10 mL 容量瓶中,加甲醇稀釋至刻度,搖勻,分別精密吸取 20 μL,按“2.3.4”項下色譜條件分析,分別進樣,記錄色譜圖,并進行積分處理。以色譜峰面積積分值(Y)為縱坐標,對照品溶液的質量濃度(X,μg/mL)為橫坐標線性回歸,得到鞣花酸的回歸方程為Y=3.08×105X-1.09×105(r=0.9996);鞣花酸在質量濃度 0.57~9.05 μg/mL 內與峰面積呈良好的線性關系。
2.3.7 精密度考察 分別吸取濃度為 45.25 μg/mL 的鞣花酸對照品溶液10 μL,按“2.3.4”項下條件重復測定6次,以色譜峰面積積分值計算相對標準偏差(RSD),結果鞣花酸峰面積的RSD(n=6)為1.60%,表明儀器精密度良好。
2.3.8 重復性考察 分別精密稱取六味石榴丸(批號:210802)樣品細粉,6 份,按“2.3.2”項下方法制備供試品溶液,按上述色譜條件測定,得鞣花酸的平均含量和 RSD 分別為 79.41 μg/g、1.19%,表明該方法的重復性良好。
2.3.9 穩定性試驗 取“2.3.2”項下同一供試品溶液,置于室溫下,分別于0 h、2 h、4 h、8 h、12 h及24 h取樣,進樣分析,計算出鞣花酸峰面積積分值的RSD為 3.00%,提示供試品溶液在 24 h 內穩定。
2.3.10 加樣回收率實驗 取已知含量的六味石榴丸(批號:210802)樣品細粉,精密稱定,共6 份,每份 0.5 g,按樣品含量100%分別加入鞣花酸對照品溶液,按“2.3.2”項下方法制備供試品溶液,依“2.3.4”項下色譜條件測定,并進行回收率的計算。測得鞣花酸的平均加樣回收率為98.14%,RSD值為1.50%。
2.3.1.10 樣品測定 精密稱取來源于不同批號的各樣品3份,每份1.0 g,按“2.3.2”項下方法制備樣品溶液,依“2.3.1.4”項下色譜條件進樣 20 μL,測定樣品中鞣花酸的含量,結果見表2。
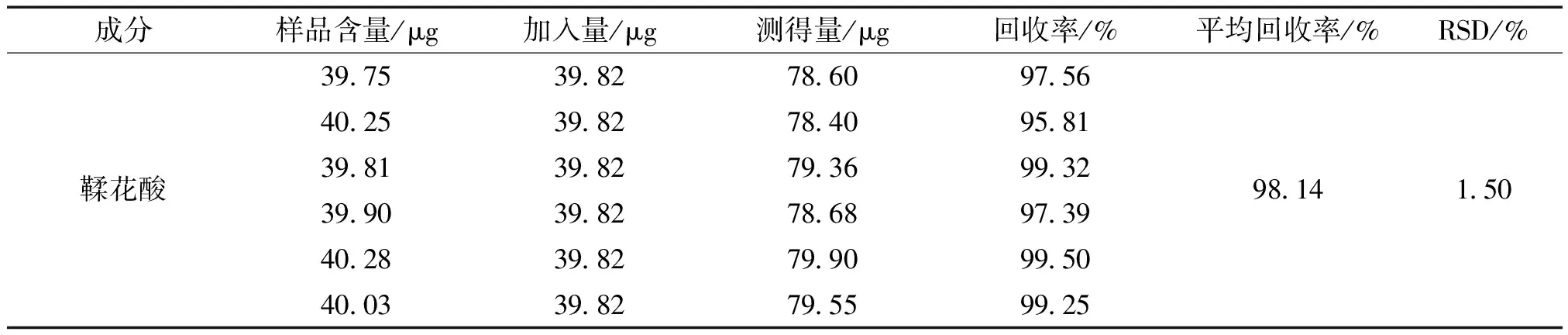
表1 加樣回收率試驗結果 (n=6)

表2 樣品中鞣花酸含量測定結果 (n=3)
2.4 六味石榴丸中羥基紅花黃色素A的HPLC含量測定
2.4.1 對照品溶液的配置 精密稱取羥基紅花黃色素A對照品 13.88 mg,置于25 mL容量瓶,用25%甲醇溶解、定容至刻線,搖勻,即得對照品儲備液,備用。
2.4.2 供試品溶液的制備 取六味石榴丸適量研細,另取約0.3 g,精密稱定,置具塞錐形瓶中,精密加入25%甲醇50 mL,稱定重量,超聲處理(功率300 W,頻率40 kHz)50 min,放冷,再稱定重量,用25%甲醇補足減失的重量,搖勻,靜置后,取上清液經0.22 μm微孔濾膜,取續濾液作為供試品溶液。
2.4.3 陰性對照樣品溶液的制備 按處方配比及制備方法制成不含紅花藥材的陰性對照樣品,按“2.4.2”項下方法制備陰性對照樣品溶液。
2.4.4 色譜條件 CAPCELL PAK-C18(250 mm×4.6 mm,5 μm);柱溫:25 ℃;以甲醇-乙腈-0.7%磷酸溶液(26∶2∶72)為流動相;流速:1.0 mL/min;檢測波長設置為403 nm。理論板數按羥基紅花黃色素A峰計算,應不得低于4000。
2.4.5 專屬性試驗 分別上述對照品溶液、供試品溶液和陰性樣品溶液,各10 μL,按“2.4.4”項下色譜條件檢測。由圖7可見,其分離度良好;陰性樣品色譜圖中,羥基紅花黃色素A對應目標化合物在相應的保留時間無色譜峰吸收出現,表明處方中其他共存成分對含量測定結果不存在相互干擾。
2.4.6 線性關系考察 分別精密移取“2.4.1”項下備用儲備液0.0626 mL、0.125 mL、0.25 mL、0.5 mL、1.0 mL、2.0 mL置 10 mL容量瓶中,加25%甲醇溶液定容至刻線,搖勻,分別精密吸取10 μL,按“2.4.4”項下色譜條件檢測,分別進樣,記錄各色譜圖。以色譜峰面積積分值(Y)為縱坐標,對照品溶液的質量濃度(X,μg/mL)為橫坐標線性回歸,結果羥基紅花黃色素A的回歸方程為Y=3.05×104X-4.91×104(r=0.9999);羥基紅花黃色素A在質量濃度 6.72~107.50 μg/mL內與峰面積呈良好的線性關系。
2.4.7 精密度考察 分別精密吸取濃度為 53.75 μg/mL 的羥基紅花黃色素A對照品溶液 10 μL,按“2.4.4”項下色譜條件重復測定6次,以色譜峰面積積分值計算其RSD值,結果羥基紅花黃色素A峰面積的RSD(n=6)為0.67%,表明儀器精密度良好。
2.4.8 重復性考察 分別精密稱取六味石榴丸(批號:210802)樣品細粉,6 份,按“2.4.2”項下方法制備供試品溶液,按上述色譜條件進樣分析,測得羥基紅花黃色素A的平均含量和 RSD值分別為5.30 mg/g、1.99%,表明該方法的重復性良好。
2.4.9 穩定性試驗 取“2.4.2”項下同一供試品溶液,室溫下放置,分別于0 h、2 h、4 h、8 h、12 h、24 h取樣,進樣測定,計算得羥基紅花黃色素A峰積分面積值的RSD 為0.84%,表明供試品溶液在24 h內穩定。
2.4.10 加樣回收率實驗 取已知含量的六味石榴丸(批號:210802)樣品細粉,精密稱定,供 6 份,每份 0.15 g,按樣品含量100%分別加入羥基紅花黃色素A對照品溶液,按“2.4.2”項下方法制備供試品溶液,依“2.4.4”項下條件,分析測定,并計算回收率。結果羥基紅花黃色素A的平均加樣回收率為99.00%,RSD值為1.34%。
2.4.11 樣品測定 精密稱取來源于不同批號的各樣品3份,每份0.3 g,按“2.4.2”項下方法制備供試品溶液,依“2.4.4”項下色譜條件分析,并測定樣品中羥基紅花黃色素A的含量,結果見表4。

表4 樣品中羥基紅花黃色素A含量測定結果 (n=3)
3 討論
3.1 顯微及TLC定性鑒別 根據文獻研究[3-4],選擇處方藥材粉末中典型顯微特征,如花粉粒的形態、石細胞的特征等作為鑒別要點,可快速對處方投料情況進行甄別。同時,建立石榴子、肉豆蔻、胡椒及紅花的TLC鑒別方法,各薄層色譜色譜條帶清晰、分離度良好。另外,在藏區走訪調研中,發現存在誤將花椒代替胡椒的情況,已建立的TLC方法可進行有效鑒別。
3.2 HPLC測定 該制劑投料中石榴子占處方組成的28.6%,其主要特征成分為鞣花酸等鞣質類物質,具有顯著的抗氧化、抗炎等活性[7],故選擇鞣花酸作為指標性成分,對石榴子的質量狀況進行考察。將鞣花酸對照品溶液,進行全波長掃描,確定252 nm為最佳檢測波長。實驗考察了乙腈-0.2%磷酸溶液(16∶84)、乙腈-0.2%磷酸溶液(18∶82)、乙腈-0.2%磷酸溶液(10∶90)等流動相體系,結果表明,以乙腈-0.2%磷酸溶液(16∶84)為洗脫系統,分離效果良好。通過對不同色譜柱溫(25 ℃、30 ℃、35 ℃)的比較,30 ℃ 時,目標峰峰形良好,基線平穩。分別對提取溶媒(25%甲醇、50%甲醇、70%甲醇、甲醇)、提取方式(熱回流、超聲提取)及提取時間(10 min、20 min、30 min、40 min、50 min、60 min)進行考察,經綜合考量最終選擇以甲醇為提取溶劑,超聲提取50 min。
六味石榴丸中紅花,具有活血通經、散瘀止痛的功效,其中羥基紅花素A是與功效相關的主要水溶性成分[13],且被《中國藥典》作為紅花的質控指標,其中所含羥基紅花素A按干燥品計應不得少于1.0%,故選擇羥基紅花素A作為指標性成分,對紅花投料的質量狀況進行考察。對羥基紅花素A對照品溶液,進行全波長掃描,確定 403 nm 為最佳檢測波長。實驗考察了甲醇-0.1%磷酸溶液(35∶65)、乙腈-0.1%磷酸溶液(35∶65)、甲醇-乙腈-0.7%磷酸溶液(26∶2∶72)等流動相體系,結果表明,以甲醇-乙腈-0.7%磷酸溶液(26∶2∶72)為最佳洗脫系統。通過對不同色譜柱溫(25 ℃、30 ℃、35 ℃)的比較,選擇25 ℃,目標峰峰形良好,基線平穩。分別對提取溶劑(10%甲醇、25%甲醇、50%甲醇、70%甲醇、甲醇)、提取方式(超聲、熱回流)及提取時間(20 min、30 min、40 min、60 min)進行考察,最終確定以25%甲醇為提取溶劑,超聲提取 50 min,進行HPLC分析樣品的制備。同時,按處方中紅花投料比折算,六味石榴丸中羥基紅花素A應不少于 1.4 mg/g。
綜上所述,本研究從顯微鑒別、TLC鑒別、含量測定等方面著手,較系統的建立了六味石榴丸的質量標準,操作方便便、專屬性強、準確可靠,可為該制劑的質量控制提供參考。
