氣相色譜-串聯質譜法同時測定三七中212種農藥殘留
2023-10-25 06:38:18王一帆吳易峰徐敦明陳達捷鄭文杰
分析測試學報 2023年10期
關鍵詞:檢測
王一帆,吳易峰,徐敦明*,陳達捷,鄭文杰,季 超
(1.福建農林大學 食品科學學院,福建 福州 350002;2.廈門海關技術中心,福建 廈門 361026;3.天津師范大學 生命科學學院,天津 300382;4.云南農業大學 云南生物資源保護與利用國家重點實驗室,云南 昆明 650091)
三七為五加科植物三七Panax notoginseng(Burk.)F.H.Chen的干燥根和根莖,具有散瘀止血、消腫止痛之功效[1],素有“南國神草”、“金不換”等美譽,產自云南、廣西等地[2-3]。三七也是我國民間最早使用的藥食兩用中藥材之一,被國家衛健委納入可用于保健食品的物品名單中[4]。近年來由于全球盛起的中藥材熱潮及其在新冠疫情中發揮的重要作用,中藥材的銷量大幅增加[5-6]。但農藥殘留問題一直阻礙著中藥材進入國際市場[7-8]。魏丹等[9]通過磁性親水親脂平衡萃取材料輔助基質固相分散萃取/高效液相色譜-串聯質譜法同時測定中藥材中的76 種農藥,在1 個金銀花樣品中檢出多菌靈。由于三七的生長周期長、性喜溫濕環境,易引起細菌滋生和病蟲害[10]。雖然三七早已采用人工種植且栽培技術不斷進步[11],但農藥不規范使用現象始終存在,難以符合市場對三七的質量要求。因此,研究三七中多農藥殘留檢測技術對于改善三七市場和控制品質有重要意義。
目前,三七的用藥以病害防治為主,蟲害防治為輔[12-13],主要涉及腐霉利、嘧菌胺、烯酰嗎啉、克菌丹等農藥。世界范圍內與三七相關的標準有美國藥典[14]、歐盟藥典[15]、韓國藥典[16]和日本藥典[17]等。我國除《中國藥典》[18]外,也有GB/T 19086-2008《地理標志產品 文山三七》[19]、T/CATCM 003-2017《無公害三七藥材及飲片的農藥殘留與重金屬及有害元素殘留限量》[20]、GB 2763-2021《食品中農藥最大殘留限量》[21]等其他相關標準。關于三七中多農殘的檢測技術有氣相色譜法(GC)及液相色譜法(LC)等。氣相色譜法適合熱不穩定性農藥,對于極性強、難揮發的農藥普遍采用液相色譜[22]。張飛[23]利用GC 法對三七中 38 種農藥殘留進行測定,結果良好。張舉成等[24]通過LC 法對三七中6種氨基甲酸酯類農藥進行檢測,均有較好的線性關系。色譜串聯質譜法具有更強的抗干擾能力、更高的靈敏度和更好的選擇性,可對定性困難、低濃度目標化合物進行測定,為基質復雜、背景干擾嚴重的中藥材農藥殘留檢測提供了技術保障[25]。Fu 等[26]采用氣相色譜-串聯質譜(GC-MS/MS)檢測了三七及其種植土壤中201 種農藥,在三七和土壤中分別檢出29 種和15 種農藥殘留,但其前處理耗時較長,不適宜日常監測。Duan 等[27]利用GC-MS/MS 測定了三七中有機氯、有機磷和擬除蟲菊酯等116 種農藥,方法靈敏度高,重復性好,但其檢測物質較少,目標組分無法涵蓋三七中可能出現的所有農藥。因此,迫切需要開發一種新的多農藥殘留快速檢測方法,以滿足中藥材三七的日常監測需求。
本研究從上述標準及GB 23200.113-2018《植物源性食品中208種農藥及其代謝物殘留量的測定 氣相色譜-質譜聯用法》[28]入手,建立了同時檢測212 種農藥殘留的GC-MS/MS 法。樣品采用FaPEx-BKT50 固相萃取(SPE)柱凈化,該柱中含有的PSA、C18、HLB、GCB 等吸附劑可有效去除三七中脂類、糖類、揮發油類以及色素等物質,很好地解決三七基質復雜、凈化難的問題,且操作簡單、可批量處理、節省時間,滿足日常監測的需求。該方法操作簡單高效,分離凈化效果好,成本低,且靈敏度高、選擇性好,可為三七藥材中多農藥殘留檢測提供參考。
1 實驗部分
1.1 儀器與設備
TQ-8040氣相色譜-三重四極桿質譜儀(配有電子轟擊(EI)源,日本島津公司);ST16R 冷凍臺式離心機(美國ThermoFisher 公司);Supelco 24 位固相萃取儀(美國Merck 公司);N-EVAP-24 氮吹儀(美國Organomation 公司);SIGMA3-18KS 高速離心機(德國Sigma 公司);MTV-100 多管旋渦混合儀(杭州奧盛儀器有限公司);MS3 圓周振蕩器(德國IKA 公司);KQ-700DE 型數控超聲波清洗器(昆山市超聲波有限公司);CPA225D電子天平(德國Sartorius公司);SF1130中藥分析研磨機(長沙中南制藥機械廠)。
1.2 材料與試劑
乙腈、乙酸乙酯、甲苯(色譜純,美國Sigma-Aldrich 公司);無水硫酸鈉(分析純,國藥集團化學試劑有限公司),中性氧化鋁柱(50 mg/3 mL,Supelco 公司),FaPEx-BKT50 SPE 柱(50 mg/3 mL,巨研科技有限公司)。212種農藥標準品名稱見表1,購自天津阿爾塔公司。
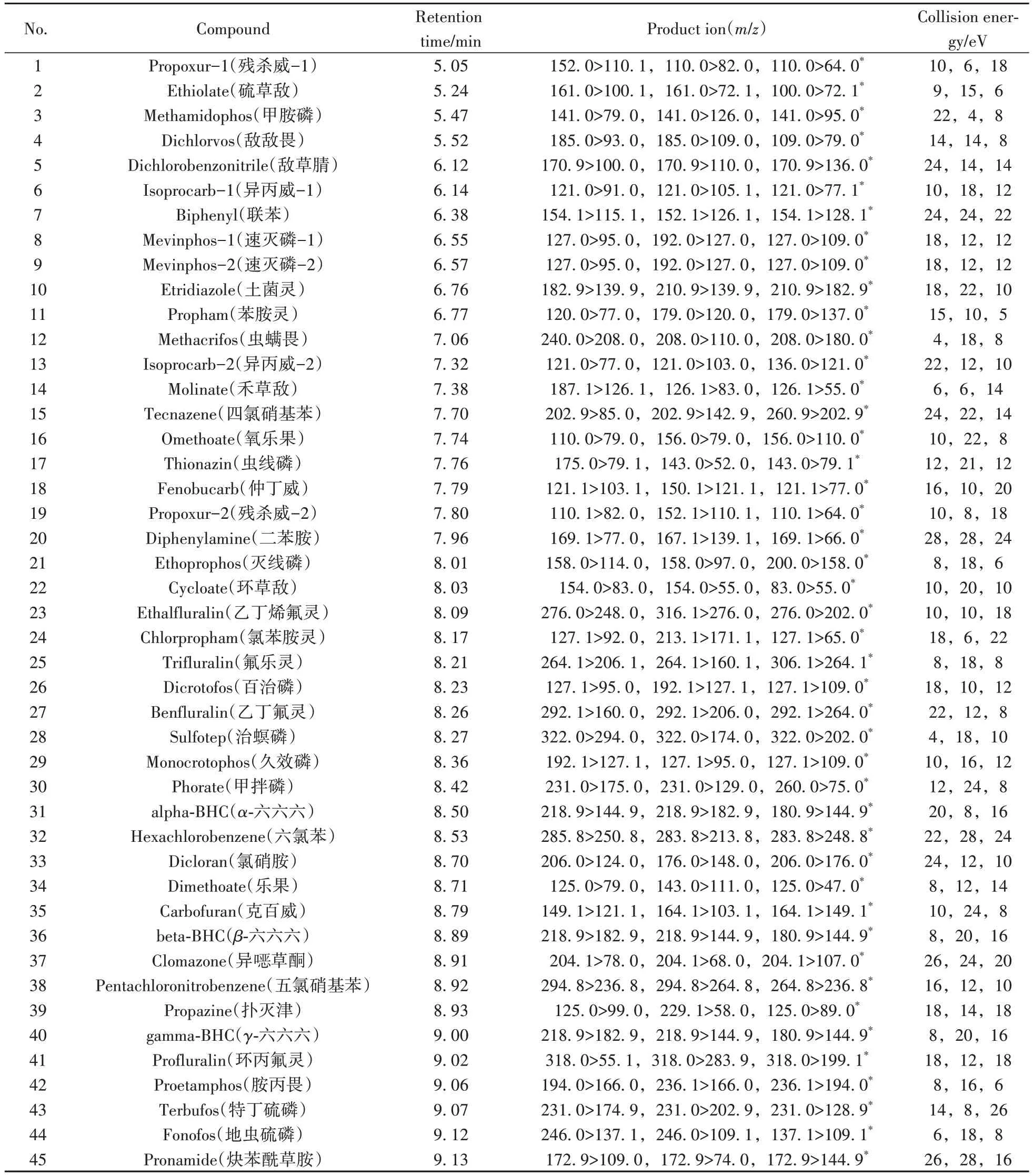
表1 212種農藥的保留時間及質譜參數Table 1 Retention times and mass spectrometry parameters of 212 pesticides
三七塊莖樣品購于市場及藥店,將其充分研磨成細粉狀,儲存在鋁箔拉鏈袋中,置于涼爽干燥的環境溫度下備用。
1.3 實驗條件
1.3.1 樣品前處理稱取5.0 g(精確至0.01 g)三七粉于50 mL 離心管中,加入15 mL 去離子水混勻,浸泡靜置30 min,加入15 mL 乙腈-乙酸乙酯(1∶3,體積比),混勻后振蕩提取20 min,加入6 g 無水硫酸鈉,4 ℃ 下8 000 r/min 離心5 min,取上清液待凈化。
用6 mL 乙酸乙酯潤洗FaPEx-BKT50 SPE 柱,取10 mL 上述上清液過柱,收集流出液,用10 mL 乙腈-甲苯(3∶1,體積比)洗脫SPE 柱,收集洗脫液緩慢氮吹近干,用1 mL 乙酸乙酯定容,振蕩混勻裝瓶待用。
1.3.2 色譜條件色譜柱:Restek Rxi-5silMS 柱(30 m × 0.25 mm,0.25 μm);升溫程序:60 ℃保持1 min,以25 ℃/min 加熱至180 ℃,再以10 ℃/min 加熱至310 ℃并保持10 min;載氣:氦氣(He),流速為1.2 mL/min;進樣量:1 μL;進樣方式:不分流進樣,吹掃時間為1 min。
1.3.3 質譜條件電子轟擊離子源(EI);電離能量:70 eV;傳輸線溫度:300 ℃;離子源溫度:230 ℃;離子監測模式:多反應監測模式(MRM);212種農藥的保留時間、定性離子、定量離子和碰撞能量等參數見表1。

(續表1)
2 結果與討論
2.1 樣品前處理條件的優化
2.1.1 提取溶劑的優化目前三七的提取溶劑主要有乙腈、0.1%乙酸-乙腈、1%乙酸-乙腈等。乙腈是農藥殘留的常用提取溶劑[29]。在乙腈中加入乙酸乙酯不僅可提高非極性農藥的提取效率,而且有助于兩相體系的形成,在一定程度上利于去除三七中的極性干擾成分。以三七中常見的8 種不同極性農藥為代表,分別考察了乙腈、乙腈-乙酸乙酯(1∶1)、乙腈-乙酸乙酯(1∶3)和乙腈-乙酸乙酯(3∶1)4種提取體系的提取效果(見圖1)。結果表明,在 100 μg/kg添加水平下,4種提取體系的提取效果差異較明顯,相對于純乙腈,乙腈-乙酸乙酯可使目標組分的回收率普遍提高,當提取溶劑為乙腈-乙酸乙酯(1∶3)時,目標組分的回收率最好。因此,選用乙腈-乙酸乙酯(1∶3)作為提取溶劑。

圖1 不同提取溶劑的提取效果對比Fig.1 Comparison on extraction effects of pesticides with different extractants
2.1.2 其它條件的選擇三七的含水量低,為保證最佳提取效果,通常會減少樣品量并添加水溶液進行浸泡[30]。目前大多數的研究表明最佳浸泡時間為30 min[31]。因此本研究選取浸泡時間為30 min。中藥材的主要提取方式有超聲提取[32]和振蕩提取等。目前大多數研究選用振蕩提取。因此本研究采用振蕩提取方式。
2.1.3 凈化條件的選擇在用無水硫酸鈉清除水分的條件下,分別考察了中性氧化鋁小柱和FaPEx-BKT50 SPE柱對三七中8種代表性農藥的凈化效果。FaPEx-BKT50 SPE柱中含有多種吸附劑,其中PSA可去除基質中的有機酸、糖類等極性干擾物;C18可有效去除基質中的脂類、固醇等非極性干擾物;HLB 可去除基質中的部分葉綠素、磷脂和揮發油;GCB 可有效去除基質中的色素。結果表明,FaPEx-BKT50 SPE 柱中的PSA、C18、GCB 以及HLB 等組分對三七的凈化效果更強,顏色去除更明顯,且目標組分的回收率相對較高。考慮到連續大量進樣會對色譜柱和儀器造成損害及污染,最終采用FaPEx-BKT50 SPE柱作為凈化條件。
2.1.4 洗脫試劑的選擇分別考察了乙酸乙酯、乙腈-甲苯(3∶1)和乙腈-乙酸乙酯(1∶3)作為洗脫試劑時對8種代表性農藥提取效果的影響。結果表明,乙腈-甲苯(3∶1)作為洗脫試劑時的提取效果最好,因此選擇乙腈-甲苯(3∶1)作為洗脫試劑。
2.1.5 洗脫體積的選擇考察了洗脫體積分別為4、6、10 mL 時對8 種代表性農藥提取效果的影響。結果表明,洗脫體積為10 mL時的提取效果最好。因此,選擇洗脫體積為10 mL。
2.2 質譜條件的優化
根據GC-MS/MS母離子和子離子一一對應的MRM 模式,通過設定多個時間段和掃描通道同時分析多種農藥成分,先通過GC對其進行分離和全掃描,確定農藥的出峰時間和一級碎片離子。選擇離子強度高的一級碎片離子作為母離子,應用電子轟擊掃描模式對母離子在不同碰撞能量下進行二次電離,選擇信號較強的二級碎片離子作為子離子,以產生信號強度最強的碰撞能量作為最優碰撞能量,優化后的條件見表1。在此條件下,212種農藥的總離子流圖(TIC)如圖2。
2.3 基質效應評價
基質效應(ME)是痕量目標物質譜分析的常見現象。采用下式計算每種化合物的基質效應:ME=[(基質匹配標準曲線的斜率/溶劑標準曲線的斜率)-1]× 100%。結果顯示,僅有18 種農藥的基質效應不明顯(<20%),如滅蟻靈、三氯殺螨砜、甲氰菊酯、β-硫丹等;92 種農藥產生中等的基質抑制效應(20%~50%),如殘殺威、益棉磷、氯苯嘧啶醇、敵草胺等;102 種農藥產生較強的基質抑制效應(≥50%),如噠嗪硫磷、苯醚菊酯、氯菊酯、溴氰菊酯、喹禾靈等。為確保實驗結果的準確性,采用基質匹配標準曲線對目標化合物進行定量分析。
2.4 方法性能評價
在10~200 μg/L范圍內,根據不同定量下限將基質混合標準溶液配制成一系列混合標準溶液。精確吸取一定量該溶液,用乙酸乙酯逐級稀釋成質量濃度為0.01、0.02、0.05、0.10、0.20 mg/L的標準工作溶液。空白基質溶液氮氣吹干后,分別加入1 mL上述標準工作溶液復溶,得到系列基質混合標準工作溶液,采用本方法測定。以進樣質量濃度為橫坐標,對應峰面積為縱坐標繪制標準工作曲線。結果表明,212種農藥在10~200 μg/L范圍內具有良好的線性關系,相關系數(r2)均大于0.999,檢出限(S/N=3,LOD)和定量下限(S/N=10,LOQ)分別為0.003~0.02 mg/kg和0.01~0.05 mg/kg(見表2)。

表2 212種農藥的相關系數、檢出限、定量下限、加標回收率及相對標準偏差(n=6)Table 2 Correlation coefficients(r2),LODs,LOQs,spiked recoveries and RSDs of 212 pesticide residues(n=6)
目前,國內外關于三七的農藥最大殘留限量(MRL)標準主要以藥典為主。《中國藥典》規定了33 種禁限用農藥的MRL值;《香港中藥材標準》(10th)[33]對20種農藥進行了規定,其MRL值為0.05~1.00 mg/kg;《臺灣藥典》(3th)[34]、《歐盟藥典》(10th)、《美國藥典》(USP44)、《韓國藥典》(10th)和《日本藥典》(18th)規定了與三七同科的人參中農藥的MRL 值。本研究中212 種農藥的LOQ 為0.01~0.05 mg/kg,均低于中國(0.01~5 mg/kg)、歐盟(0.01~4 mg/kg)、美國(0.01~4 mg/kg)、韓國(0.01~5 mg/kg)和日本(0.01~75 mg/kg)建立的MRL值。
2.5 回收率與相對標準偏差
在空白樣品中準確添加0.02、0.04、0.1 mg/kg 三水平的農藥混合標準溶液,每個濃度做6次平行實驗(見表2)。結果表明,212 種農藥的平均回收率為60.0%~121%,相對標準偏差(RSD)為0.90%~31%。
2.6 實際樣品分析
為驗證本方法的實用性并了解市售三七中農藥殘留情況,采用本方法對采自藥店、藥材市場及種植地等的66批三七藥材進行了檢測。結果表明,66批樣品中均檢出多種農藥。從農藥種類來看,共檢出40 種農藥,以腐霉利、毒死蜱、六氯苯、嘧菌胺和烯酰嗎啉等檢出最多,檢出率分別為79.3%、72.1%、75.8%、62.5%、60.9%,其中毒死蜱的最高殘留量達324 μg/kg,腐霉利的最高殘留量達64μg/kg,烯酰嗎啉的最高殘留量達1 023 μg/kg。從種植地區來看,所有林下種植的三七農藥殘留普遍較少,每批檢出農藥在5 ~ 10種之間;農田種植的三七農藥殘留檢出較多,其中62%的樣品檢出13~25種農藥。整體來看,雖然林下種植的三七農藥殘留相對較少,但無論是農田種植或是林下種植,三七中毒死蜱、腐霉利、六氯苯的檢出率均較高,應引起重點關注和防控,從而提升中藥材三七的品質。
3 結 論
本研究建立了GC-MS/MS 同時測定三七中212 種農藥的檢測方法。針對三七色素多、基質雜的特點,選用FaPEx-BKT50固相萃取柱進行前處理,除雜效果好,凈化更徹底。本方法靈敏度高,準確性好,一次性前處理可同時檢測多種農藥,為中藥材三七的農殘檢測提供了一種切實可行的方法。
猜你喜歡
中國設備工程(2022年12期)2022-07-11 04:33:00
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:36
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:34
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:50
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:48
