牛奶中69種獸藥殘留的超高效液相色譜-四極桿-飛行時間質(zhì)譜快速篩查
2023-10-25 06:38:26嚴祖浩李曉薇
分析測試學(xué)報 2023年10期
嚴祖浩,李曉薇,夏 曦
(中國農(nóng)業(yè)大學(xué) 動物醫(yī)學(xué)院,北京 100193)
牛奶已經(jīng)成為我國消費者膳食的重要組成部分,其主要營養(yǎng)成分包括蛋白質(zhì)(3.0%~3.9%)、乳糖(4.4%~5.6%)、脂肪(3.3%~5.4%)和礦物質(zhì)(0.7%~0.8%)[1]。此外,牛奶還是多種微量營養(yǎng)元素的重要來源,包括鈣、磷、鎂、鋅、碘、鉀、維生素 A、維生素D、維生素 B12,這些微量元素對兒童和青少年的生長發(fā)育,以及維持成人身體健康均具有極大幫助[2-3]。由于養(yǎng)殖場對抗菌藥物的不合理和不規(guī)范使用,導(dǎo)致牛奶中抗菌藥物殘留超標并嚴重危害人類公共安全[4]。β-受體激動劑是苯乙醇胺類化合物,在動物飼養(yǎng)中能夠改變養(yǎng)分的代謝途徑,促進動物肌肉生長及骨骼肌蛋白質(zhì)的合成,加速脂肪轉(zhuǎn)化和分解,提高牲畜瘦肉率[5]。但畜產(chǎn)品中的激動劑殘留可能會造成人體健康風(fēng)險,哮喘或心血管疾病患者攝入后還可能引起頭暈、心悸、血壓升高、心率加快等心血管系統(tǒng)的中毒反應(yīng),甚至導(dǎo)致死亡[6]。為了確保消費者的食品安全,不同國家對牛奶中抗菌藥物的最大殘留限量(MRLs)做出了規(guī)定,我國在GB 31650-2019《食品安全國家標準 食品中獸藥最大殘留限量》中規(guī)定[7],除磺胺二甲嘧啶(25μg·kg-1)外的磺胺類藥物的MRL 均為100 μg·kg-1;土霉素、金霉素、四環(huán)素的MRL 均為100 μg·kg-1。世界上大多數(shù)國家和地區(qū),如歐盟、日本等已禁止β-受體激動劑類藥物用于食品動物,我國農(nóng)業(yè)農(nóng)村部第250號公告規(guī)定禁止使用β-受體激動劑類藥物作為獸藥和飼料添加劑[8]。
目前,牛奶中多種獸藥殘留的儀器檢測方法國內(nèi)外均有報道,包括毛細管電泳法[9-10]、高效液相色譜法[11-12]、液相色譜-串聯(lián)質(zhì)譜法[13-14]、超高效液相色譜-飛行時間質(zhì)譜法[15-16]等。樣品前處理是檢測牛奶中獸藥殘留的關(guān)鍵步驟,一般包括液液萃取[17]、常規(guī)固相萃取[18-19]、分散固相萃取[13,20-21]等。本實驗旨在建立基于超高效液相色譜-四極桿-飛行時間質(zhì)譜(UPLC-Q-TOF MS)的牛奶中多種獸藥殘留的篩查方法,為牛奶中的獸藥殘留檢測提供參考。
1 實驗部分
1.1 試劑與材料
乙腈(色譜純,賽默飛世爾科技有限公司;質(zhì)譜純,上海安譜實驗科技股份有限公司),甲醇、正己烷(質(zhì)譜純,上海安譜實驗科技股份有限公司),甲酸(質(zhì)譜純,奇睿化學(xué)科技有限公司),實驗用水由Millipore超純水儀制備。
以下為標準品:
1種喹諾酮類藥物內(nèi)標:恩諾沙星-D5。
30 種β-受體激動劑類藥物:班布特羅、巴美生、溴布特羅、溴代克侖特羅、卡布特羅、西布特羅、克侖塞羅、克侖西羅、鹽酸克侖西羅、異克侖潘特、鹽酸克侖特羅、克侖丙羅、氯丙那林、非諾特羅、福莫特洛、羥甲基克侖特羅、鹽酸苯氧丙酚胺、拉貝洛爾、苯乙醇胺A、利妥特靈、索他洛爾、特布他林、妥布特羅、齊帕特羅、馬噴特羅、馬布臺諾、吡布特羅、可爾特羅、奧西那林、沙美特羅。
15 種β-受體激動劑類藥物內(nèi)標:溴布特羅-D9、卡布特羅-D9、西布特羅-D9、鹽酸克侖特羅-D5、克侖丙羅-D7、氯丙那林-D7、鹽酸氧丙酚胺-13C6、沙美特羅-D(3Salmeterol-D3)、齊帕特羅-D7、苯乙醇胺A-D3、特布他林-D9、妥布特羅-D9、馬布臺諾-D9、吡布特羅-D9、奧西那林-D7。
4種四環(huán)素類藥物:四環(huán)素、金霉素、土霉素、多西環(huán)素。
β-受體激動劑類藥物及內(nèi)標由德國BVL 實驗室提供,磺胺類、四環(huán)素類和喹諾酮類藥物由天津阿爾塔公司提供。
1.2 儀器與設(shè)備
超高效液相色譜儀(ACQUITY UPLC I-Class PLUS,沃特世公司),四極桿-飛行時間質(zhì)譜儀(X500 B,美國SCIEX公司),高速冷凍離心機(Eppendorf公司),TARGIN VX-Ⅲ多管渦旋振蕩器(北京踏錦科技有限公司),N-EVAP-112氮吹儀(美國Organomation公司)。
1.3 溶液配制
標準溶液制備:分別稱取10 mg 標準品于10 mL 容量瓶中,用甲醇溶解定容,配制成質(zhì)量濃度為1 mg·mL-1的單標儲備液,置于-20 ℃冰箱中避光保存。
混合標準工作液:根據(jù)實際需要,將單標儲備液混合配制成不同質(zhì)量濃度的混合標準工作液,置于-20 ℃冰箱中保存。
1.4 儀器方法
1.4.1 色譜條件色譜柱:ACQUITY BEH C18(2.1 mm × 50 mm,1.7 μm);柱溫40 ℃;進樣體積2μL;流速:0.3 mL/min;流動相A 相為0.1%甲酸溶液,B 相為含0.1%甲酸的甲醇-乙腈(2∶8,體積比,下同)溶液,梯度洗脫程序:0~2 min,95%~85% A;2~5 min,85%~60% A;5~7 min,60%~5% A;7~8.5 min,5% A;8.5~8.6 min,5%~95% A;8.6~10 min,95% A。
1.4.2 質(zhì)譜條件電噴霧正離子模式(ESI+);霧化氣:344 737.85 Pa(50 psi);加熱氣:344 737.85 Pa(50 psi);氣簾氣:241 316.50 Pa(35 psi);加熱氣溫度:500 ℃;碰撞氣:48 263.30 Pa(7 psi);電噴霧電壓:5 500 V;TOF MS 一級母離子掃描參數(shù):掃描范圍100~10 000 Da;去簇電壓:80 V;TOF MS/MS 子離子掃描參數(shù):掃描范圍:50~1 000 Da;去簇電壓:80 V;碰撞能量:35 V;碰撞電壓的變化步階:15 V。
使用SCIEX公司提供的正模式調(diào)諧溶液,對儀器質(zhì)量軸進行校準,參比離子為m/z132.904 90、m/z266.159 81、m/z354.212 24、m/z442.264 67、m/z609.280 66、m/z829.539 33、m/z961.436 96、m/z1 446.732 24 和m/z1 561.603 32,子離子參比離子為m/z58.065 1、m/z86.096 4、m/z235.135 6、m/z174.091 3、m/z365.186 0、m/z448.196 6、m/z724.496 7、m/z607.417 8、m/z512.344 3、m/z811.528 8。
1.5 樣品前處理
稱取1.00 g 牛奶,添加內(nèi)標后靜置10 min,加入0.5%甲酸乙腈5 mL,渦旋振蕩3 min,4 ℃下10 000 r/min 離心5 min。取上清液,加入10 mL 乙腈飽和的正己烷,手搖20 次,4 ℃下10 000 r/min 離心5 min,棄上清液。下層提取液40 ℃氮氣吹干,加入0.1%甲酸甲醇300 μL,渦旋1 min,加入0.1%甲酸溶液700 μL,渦旋1 min,再加入5 mL 乙腈飽和的正己烷,手搖20 次,4 ℃下10 000 r/min 離心5 min,棄上清液。下層溶液于4 ℃下14 000 r/min 離心20 min,過0.22 μm濾膜,上機檢測。
2 結(jié)果與討論
2.1 色譜條件的優(yōu)化
本實驗對比了不同色譜柱和流動相對藥物的分離能力,考察了不同色譜柱(COPTECS UPLC Shield RP18(2.1 mm × 100 mm,1.6 μm)、ACQUITY UPLC BEH C18(2.1 mm × 50 mm,1.7 μm))和流動相(0.1%甲酸-乙腈和0.1%甲酸-甲醇/乙腈(2∶8))對69種獸藥分離、離子化效率和峰形的影響。結(jié)果表明:Shield RP18 對磺胺對甲氧嘧啶和磺胺甲氧噠嗪的分離度差,而BEH C18對上述同分異構(gòu)體的分離度良好(圖1);與0.1%甲酸-乙腈相比,有機相中增加甲醇可以獲得更好的分離效果[22],且含0.1%甲酸的甲醇-乙腈(2∶8)作為有機相可使目標化合物的峰形更加對稱。所以最終使用BEH C18(2.1 mm ×50 mm,1.7 μm)色譜柱,0.1%甲酸為水相,含0.1%甲酸的甲醇-乙腈(2∶8)為有機相進行梯度洗脫,以實現(xiàn)目標物化合物的有效分離。69種獸藥的提取離子色譜圖見圖2。
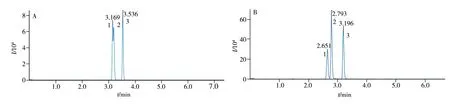
圖1 不同色譜柱的分離效果Fig.1 Separation effect of different columns

圖2 69種獸藥基質(zhì)標準溶液的總離子流色譜圖Fig.2 Total ion chromatogram of the matrix standard solution of 69 veterinary drugs
2.2 質(zhì)譜條件的優(yōu)化
高分辨質(zhì)譜的數(shù)據(jù)采集模式可分為數(shù)據(jù)依賴采集模式(DDA)和數(shù)據(jù)非依賴采集模式(DIA)兩種,其中DDA 是根據(jù)提前設(shè)定的條件,如離子強度、信噪比等對母離子進行篩選,獲得二級質(zhì)譜信息,所得到的二級譜圖無干擾離子,但在非靶向篩查時會出現(xiàn)分析重復(fù)性較差,二級信息覆蓋率低等問題;DIA 則不預(yù)先對母離子進行選擇,在低能量時獲得一級質(zhì)譜圖,高能量時獲得二級碎片,理論上能夠全面地獲得所有離子的碎片信息,但所獲的二級譜圖存在其他母離子碎片的干擾,且數(shù)據(jù)解析時間更長[23]。本實驗對比了DDA和DIA兩種模式在69種獸藥篩查中的差異,發(fā)現(xiàn)二者在目標化合物定量方面的差異不顯著;DDA 模式的二級質(zhì)譜干擾離子較少,可獲得含較高質(zhì)量碎片信息的二級圖譜,以磺胺噻唑為例,其DDA 和DIA 模式下的二級質(zhì)譜圖見圖3。將所有目標化合物通過超高效液相色譜儀進行色譜分離,用Q-TOF MS 進行質(zhì)譜信息采集,并在m/z100~1 000 范圍內(nèi)進行一級質(zhì)譜全掃描,采用DDA模式進行二級質(zhì)譜掃描分析,收集69種目標化合物的保留時間、母離子和子離子精確分子質(zhì)量信息,結(jié)果見表1。

表1 69種獸藥的質(zhì)譜參數(shù)Table 1 Mass spectrometry parameters of 69 veterinary drugs
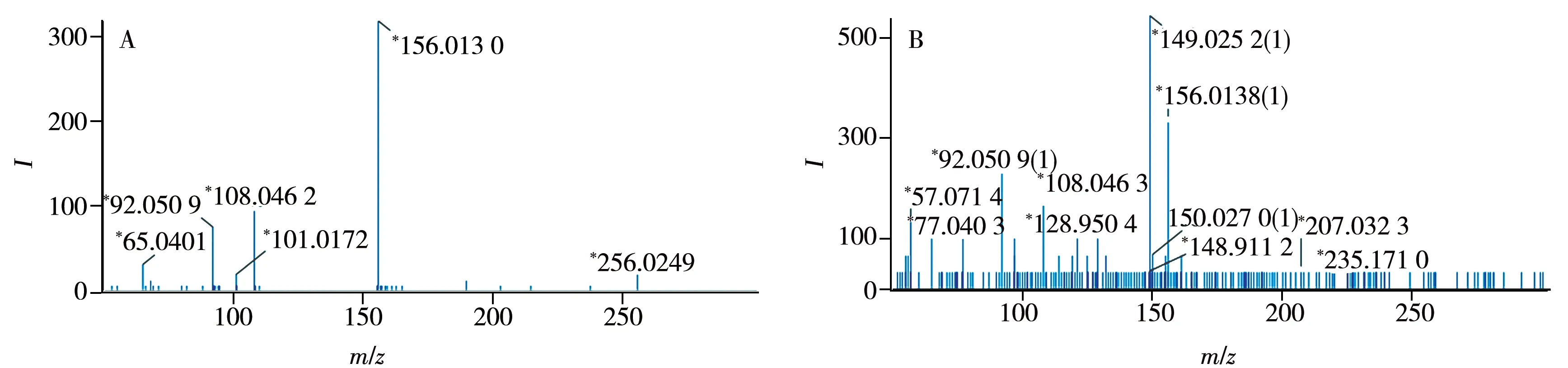
圖3 磺胺噻唑的二級質(zhì)譜圖Fig.3 MS/MS spectra of sulfathiazole
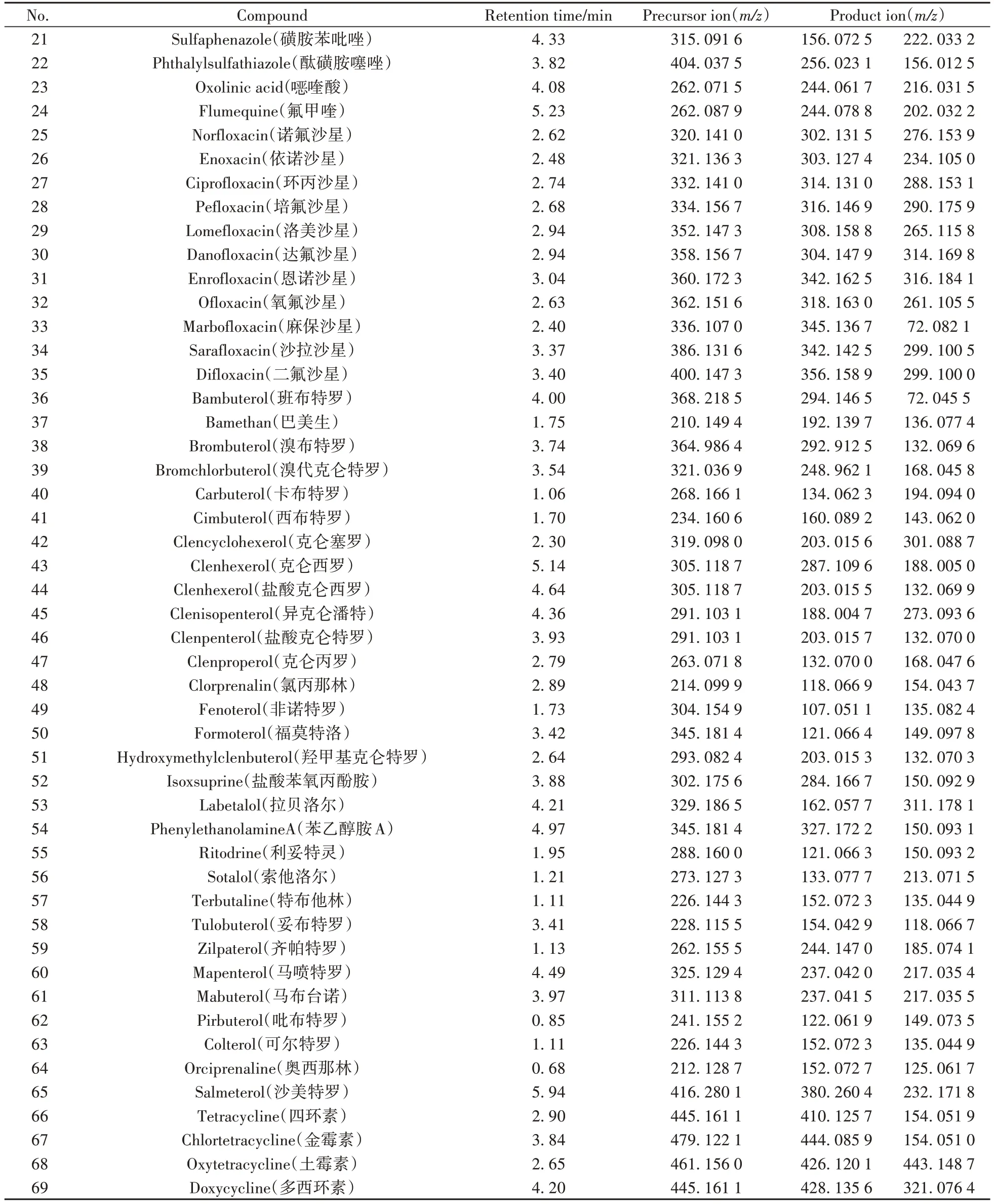
(續(xù)表1)
2.3 前處理條件的優(yōu)化
牛奶中含有豐富的蛋白質(zhì),可能會干擾目標化合物的檢測以及導(dǎo)致色譜柱或儀器管路堵塞等問題,因此需要進行蛋白沉淀。由于對不同種類藥物同時提取存在一定的困難,文獻顯示,對于本文所選的69 種目標物,酸性提取液的效果優(yōu)于中性和堿性提取液[22,24]。本實驗考察了0.1%甲酸乙腈、0.5%甲酸乙腈和1%甲酸乙腈3 種不同提取液對目標化合物的提取效果,結(jié)果表明,隨著甲酸體積分數(shù)的增加,四環(huán)素類和磺胺類藥物的回收率增加,喹諾酮類和β-受體激動劑類藥物的回收率無明顯變化,但1%甲酸乙腈提取部分磺胺類藥物時的回收率偏高,與文獻[25]類似。實驗最終選擇0.5%甲酸乙腈作為提取試劑。
牛奶基質(zhì)復(fù)雜,除富含蛋白質(zhì)外,還有乳脂、乳糖、碳水化合物和少量礦物質(zhì)等雜質(zhì),因此有必要對樣品進行凈化,減少基質(zhì)的影響。常見的牛奶凈化方法有固相萃取法、低溫冷凍除脂法[24]和正己烷除脂法,其中固相萃取法常使用HLB 和Prime HLB 等萃取柱。本實驗以乙腈飽和的正己烷進行除脂凈化,通過提取后和復(fù)溶后兩次除脂,可有效去除牛奶中大部分的基質(zhì)干擾,減少了過柱等繁瑣操作步驟,更加經(jīng)濟快速,有利于實際樣品的高通量檢測分析。
2.4 基質(zhì)匹配標準曲線及靈敏度
取空白牛奶樣品,按“1.5”進行樣品前處理,量取混合標準工作液適量,用空白樣品提取液溶解稀釋,配制成系列基質(zhì)匹配標準工作溶液,對其中的目標化合物進行測定。對于采用外標法的化合物,以化合物的離子峰面積為縱坐標(Y),標準溶液濃度為橫坐標(X),繪制標準曲線;對于使用內(nèi)標法的化合物(13 種喹諾酮類藥物、溴布特羅、西布特羅、異克侖潘特、鹽酸克侖特羅、克侖丙羅、氯丙那林、福莫特洛、鹽酸苯氧丙酚胺、苯乙醇胺A、索他洛爾、特布他林、妥布特羅、齊帕特羅、馬布臺諾、吡布特羅、奧西那林、沙美特羅),以化合物的離子峰面積和對應(yīng)內(nèi)標峰面積的比值為縱坐標(Y),標準溶液濃度為橫坐標(X),繪制標準曲線。69種目標化合物在10~200 μg·kg-1范圍內(nèi)呈現(xiàn)良好的線性關(guān)系,相關(guān)系數(shù)(r2)均不低于0.99(表2)。以各目標化合物信噪比為10(S/N=10)時對應(yīng)的濃度作為定量下限(LOQ),69種目標物在牛奶中的LOQ為10 μg·kg-1,具有較好的靈敏度。
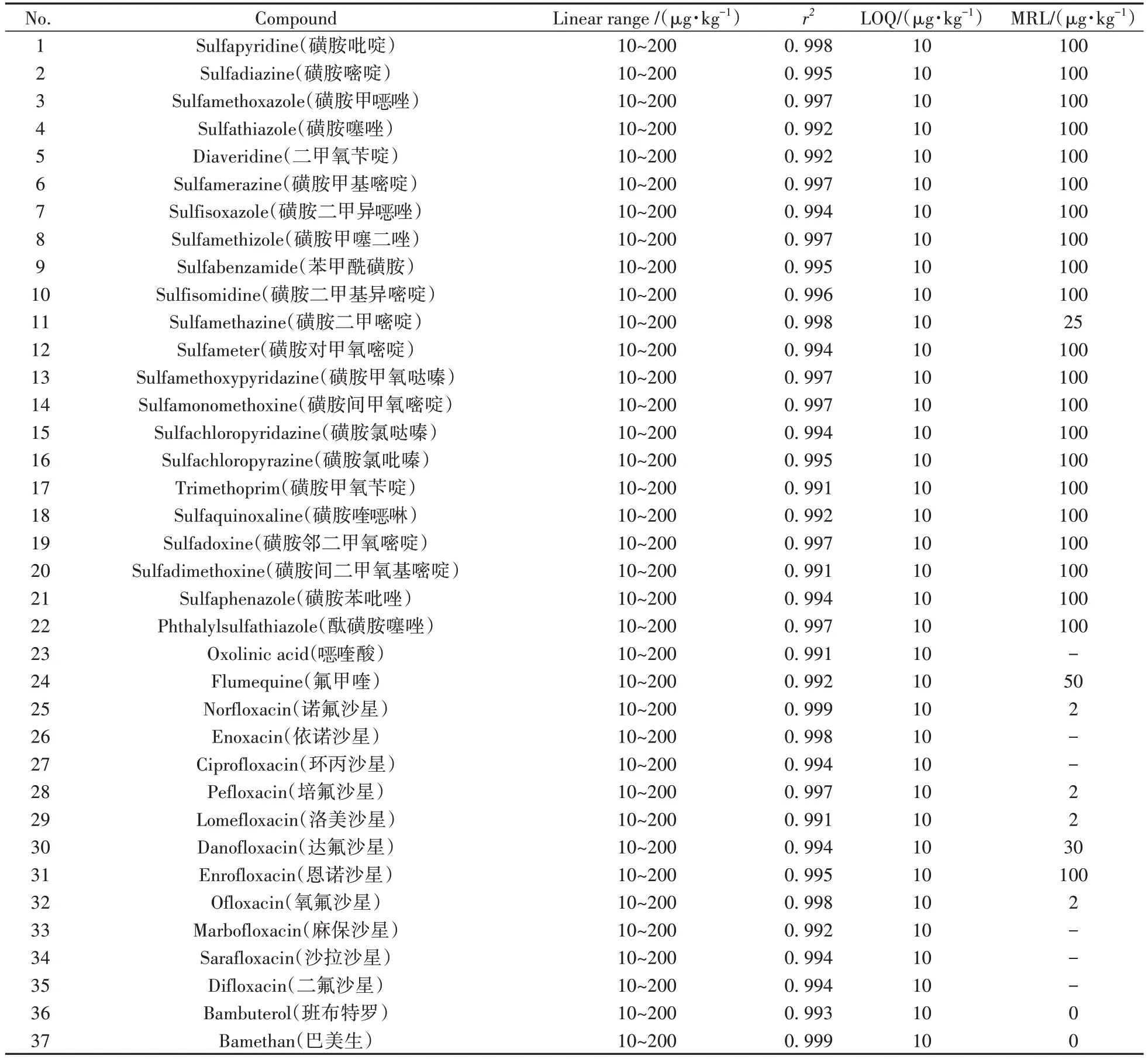
表2 69種獸藥的線性范圍、相關(guān)系數(shù)、定量下限及最高殘留限量Table 2 Linear ranges,correlation coefficients,limits of quantification and maximum residue limits for 69 veterinary drugs
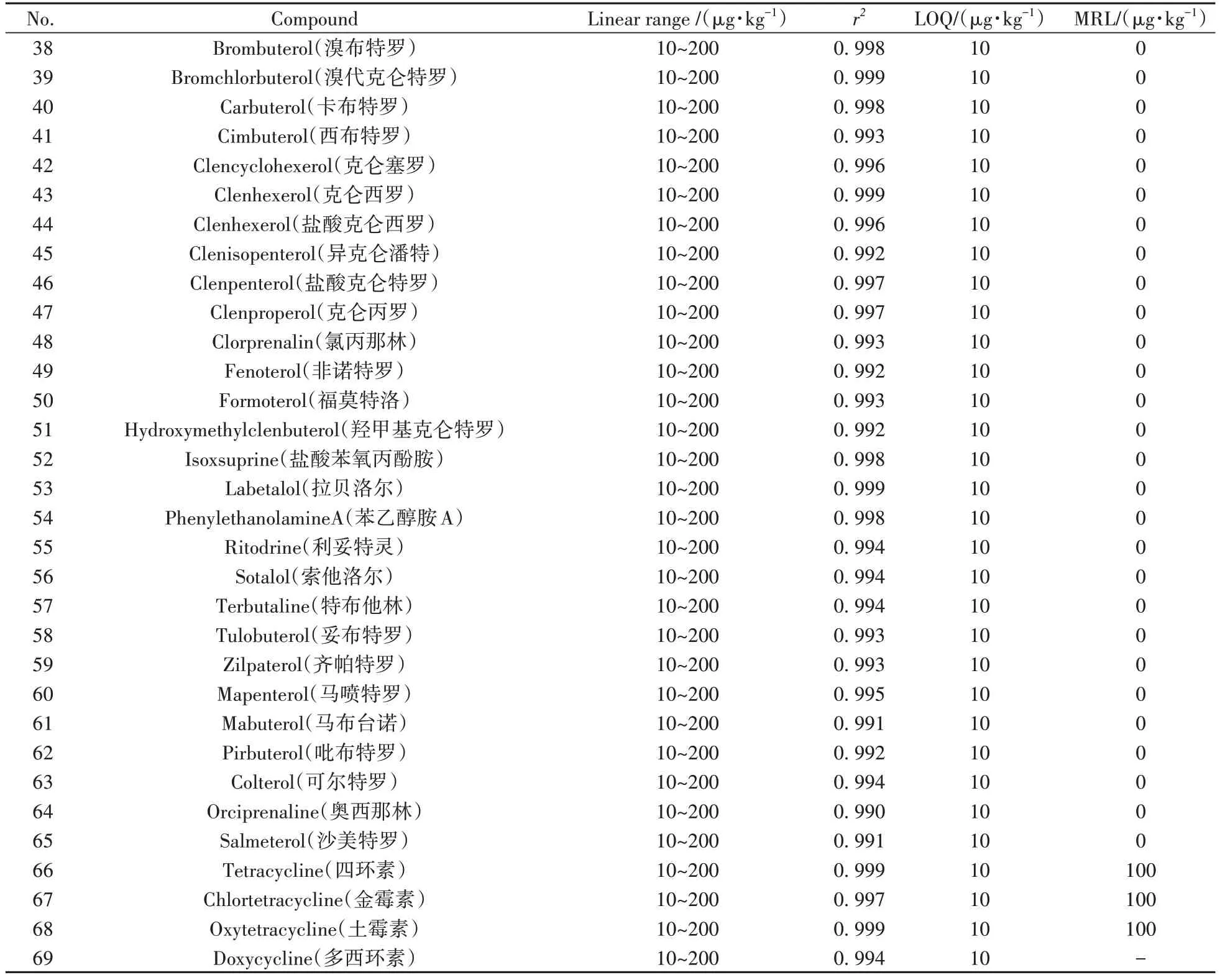
(續(xù)表2)
2.5 回收率與相對標準偏差
對牛奶樣品中的69種獸藥進行10、50、100 μg·kg-13個水平的加標,每個加標水平平行測定6次,計算批內(nèi)平均回收率和批內(nèi)相對標準偏差(RSD);連續(xù)測定3 天,計算批間平均回收率和批間RSD。外標法定量藥物的批內(nèi)平均回收率為59.0%~120%,批內(nèi)RSD不大于12%,批間平均回收率為62.0%~119%,批間RSD 不大于11%;內(nèi)標法定量藥物的批內(nèi)平均回收率為75.4%~117%,批內(nèi)RSD 不大于12%,批間平均回收率為79.3%~107%,批間RSD 不大于13%,結(jié)果見表3。加標實驗結(jié)果表明該方法具有良好的回收率和精密度,滿足多殘留篩查分析要求。
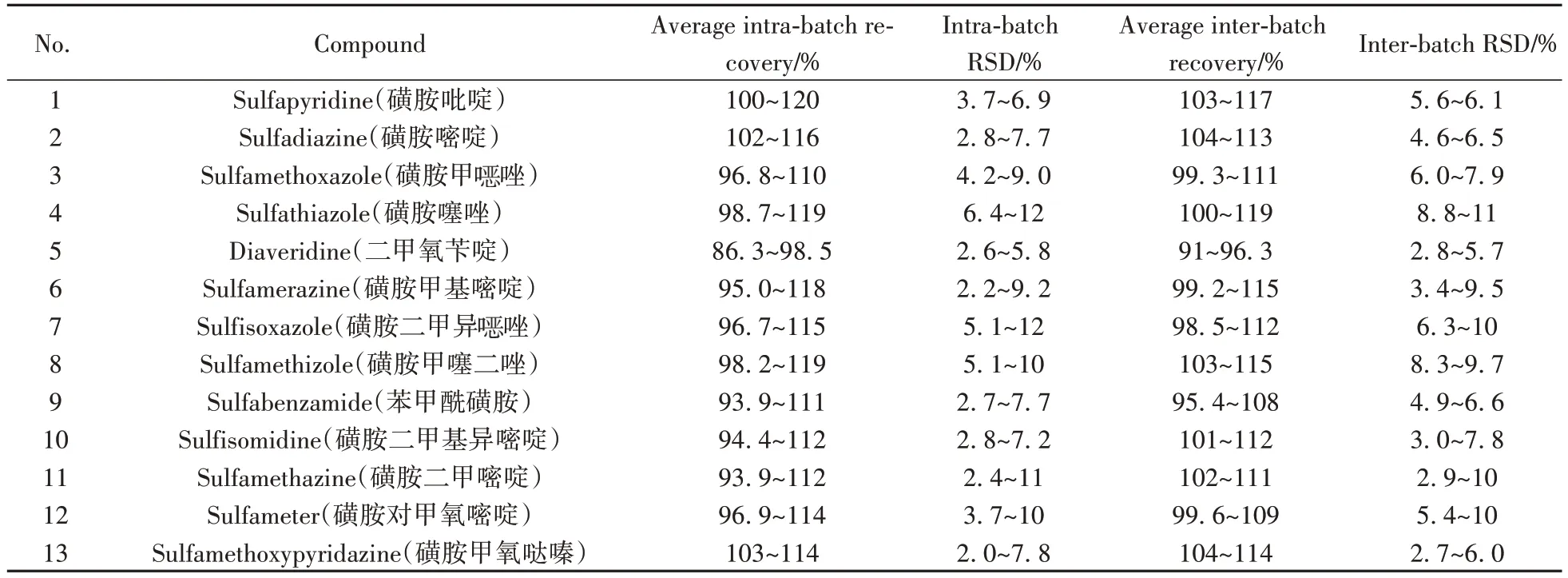
表3 牛奶中69種獸藥的加標回收率和相對標準偏差Table 3 Spiked recoveries and relative standard deviations of 69 veterinary drugs in milk
2.6 實際樣品分析
采用所建立的方法對市場采集的34種不同牛奶樣品進行篩查。首先以一級全掃描質(zhì)譜數(shù)據(jù)的母離子精確質(zhì)量數(shù)和保留時間進行比對,通過二級質(zhì)譜碎片離子對檢出的疑似陽性樣品進行確證分析。結(jié)果在1 份樣品中檢出恩諾沙星,含量為18.3 μg·kg-1;另有一份樣品檢出四環(huán)素,含量為32.6 μg·kg-1(圖4)。兩種藥物的檢出含量均未超出其最大殘留限量(100 μg·kg-1)。
3 結(jié) 論
本研究建立了超高效液相色譜-四極桿-飛行時間質(zhì)譜測定牛奶中β-受體激動劑類、磺胺類、喹諾酮類、四環(huán)素類4 類共69 種限用和禁用獸藥的篩查方法。與傳統(tǒng)獸藥殘留檢測方法相比,本方法優(yōu)化了樣品前處理步驟,可同時快速篩查牛奶中69 種獸藥的殘留量,具有操作簡單、檢測效率高等優(yōu)點。所建立的方法在快速篩查牛奶中獸藥殘留方面有明顯優(yōu)勢,可一次性分析多類不同目標化合物,為牛奶中的獸藥殘留監(jiān)管提供了有力的技術(shù)支撐。
