BALB/c突變卷毛小鼠基因組學測序差異的分析*
2023-11-02 02:07:22湯紫榮李曉娟姜棋予李興杰孫慧偉李瑞生
實驗動物科學 2023年5期
湯紫榮 李曉娟 姜棋予 李興杰 孫慧偉 李瑞生
(1.中國人民解放軍總醫院第五醫學中心住院與病案管理科,北京 100039) (2.中國人民解放軍總醫院第五醫學中心感染病醫學部研究所,北京 100039)
全基因組測序是指應用新型測序儀器測量不同機體基因組間的差異,并通過生物信息手段對與性狀關聯的遺傳變異信息及基因組結構進行分析與注釋[1]。測序技術已經由基于雙脫氧末端終止法的 Sanger測序技術[2]發展到現在的第二代、第三代測序技術[3],實現了從低讀長到超高讀長、從光學檢測到電子傳導檢測的雙重跨越測序技術,在動物基因組測序發展中起了非常重要的作用[4]。BALB/c突變卷毛鼠是本實驗室發現的突變小鼠,經近交培育已成為較為成熟、遺傳穩定的近交系突變卷毛小鼠品系[5]。前期對其外觀特征以及血液學指標進行了基礎性探討研究[6-7],尤其應用微衛星位點標記對BALB/c突變卷毛鼠進行檢測分析,發現該小鼠存在較高的突變率,且與無毛小鼠突變是兩個完全不同的突變系[8]。因此本實驗利用Illumina PE 150對BALB/c突變卷毛鼠與BALB/c小鼠進行測序,并進行生物信息學分析,以期全面了解突變鼠與BALB/c小鼠之間的基因組差異性,為今后更好地開發和利用該鼠作為優良的動物模型提供可靠的生物學信息基礎。
1 材料和方法
1.1 實驗動物
正常BALB/c小鼠和BALB/c突變卷毛鼠各1只,SPF級,雌性,6周齡。正常BALB/c小鼠來自突變群體中的正常小鼠,BALB/c卷毛鼠來自于本實驗室近交培育,生產許可證號【SCXK(軍)2012-0004】。動物飼養在中國人民解放軍總醫院第五醫學中心動物實驗中心,使用許可證號【SYXK(軍)2017-0016】。本實驗通過了動物倫理委員會審查,倫理審批號:IACUC-2017-006。
1.2 實驗方法
1.2.1動物組織采集和DNA提取:從BALB/c小鼠和BALB/c突變卷毛鼠后肢分別取一小塊肌肉,立即用PBS或0.9%氯化鈉溶液漂洗,去除血漬,并剔除結締組織和脂肪組織,吸干水分;將肌肉塊修剪成長寬高均≤0.5 cm的小塊(組織塊越小,保存效果越好);將處理好的組織樣品置于2 mL的旋蓋凍存管中,立即液氮速凍,送去北京美吉桑格生物醫藥科技有限公司。而后對2個樣本進行基因組DNA的提取、質檢和定量檢測,以及后續的建庫測序。
1.2.2文庫構建及測序:2個樣品分別構建1個Illumina PE測序文庫,共計2個Illumina 測序文庫;插入片段400 bp。Illumina PE 150每個樣品提供84 G clean data,Q30值≥80%。
1.2.3信息分析
1.2.3.1原始數據質控和過濾:測序結果得出的原始測序序列(Raw Reads)由于含有帶接頭的、低質量的Reads,因此為了提高信息分析的質量,必須對Raw Reads過濾,得到Clean Reads,后續分析都在Clean Reads的基礎上進行。對下機得到的Raw Data進行質控得到Clean Data。
1.2.3.2數據比對:進行單堿基突變(SNP)和 插入缺失(Indel)的檢測及注釋;Indel是指基因組中小片段的插入和缺失序列。利用SAMTOOLs檢測長度小于50 bp的小片段插入與缺失(Indel)。對結構變異(SV)和拷貝數變異(CNV)進行檢測和注釋。
2 結果
2.1 基因組基本信息對比結果
本次測序正常小鼠與卷毛小鼠產生的Raw Data分別為122.85 Gb和118.04 Gb,過濾后的Clean Data 分別為119.82 Gb和112.18 Gb,GC含量分別為40.63%和40.48%,表明測序質量合格。GC分布也正常,說明建庫測序成功(表1)。正常小鼠與卷毛小鼠的基因組覆蓋深度統計(表2)。

表1 測序質量統計表Table 1 Sequencing quality statistics

表2 樣品覆蓋深度和覆蓋度統計Table 2 Sample coverage depth and coverage statistics
2.2 遺傳變異檢測對比分析
卷毛小鼠的雜合分型SNPs總數顯著高于正常小鼠,而其他數值卻沒有顯著差異(表3),同時正常小鼠與卷毛小鼠之間的累計SNPs深度分布也存在顯著的差異(圖1)。卷毛小鼠與正常小鼠插入缺失(Indels)以及累計Indels深度分布均存在差異(圖2,3)。卷毛小鼠與正常小鼠的SV類型中大片段的插入卷毛小鼠大于正常小鼠,而其他類型則小于正常小鼠(表4)。而拷貝數變異(CNVs)結果顯示,正常小鼠和卷毛小鼠的拷貝數增加的個數分別為1 270和967個;而拷貝數減少的個數分別為5 027和3 505個。
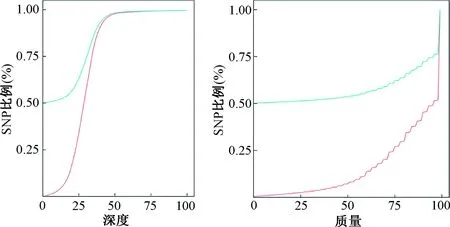
注:紅線為卷毛小鼠;藍線為正常小鼠。Note:The red line is curly mice; The blue line is normal mice.圖1 累積SNP深度分布Fig.1 Cumulative SNP depth distribution
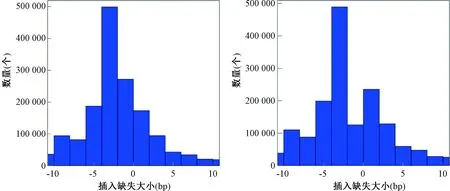
注:A.卷毛小鼠 ;B.正常小鼠。Note:A. Curly mouse; B. Normal mouse.圖2 插入缺失(Indel)分布情況Fig.2 Distribution of insertion and deletion
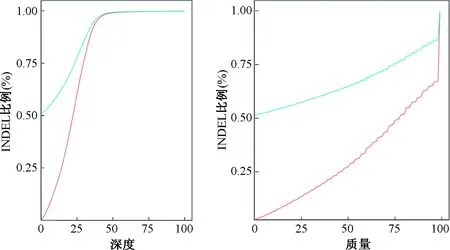
注:紅線為卷毛小鼠;藍線為正常小鼠。Note. The red line is curly mice; The blue line is normal mice.圖3 累積Indel深度分布Fig.3 Cumulative Indel depth distribution
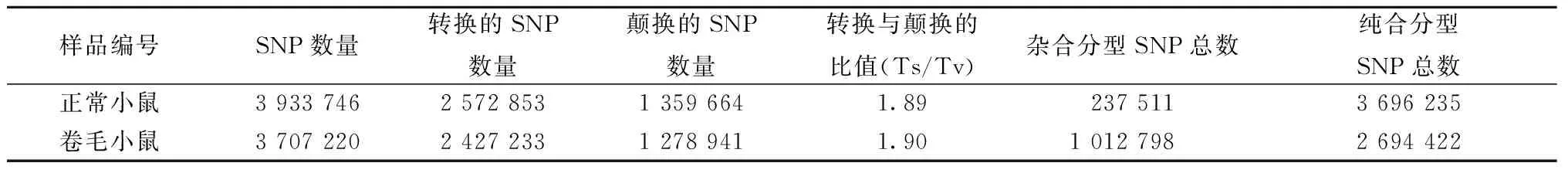
表3 SNP數據統計表(個)Table 3 SNP data statistics(個)

表4 SV預測結果統計表Table 4 Statistical table of SV prediction results
3 討論
全基因組重測序的特點是具有分析結果快速、準確、靈敏度高和自動化,指在參考已知基因組序列的物種信息基礎上,對不同個體整個基因組進行測序,再通過生物信息學分析及在個體或群體水平進行序列差異性分析[9]。那么在測序過程中可以檢測到大量與性狀關聯的遺傳變異信息(包括SNP、Indel、SV和CNV甚至新基因等)。研究人員利用全基因組測序方法比較近交系小鼠C57BL/10和C57BL/6的基因組差異,篩選出 4 個小鼠免疫應答差異基因[10]。研究人員完成了美洲大蠊的全基因組測序,也是大蠊屬Periplaneta 昆蟲的第一個基因組,為美洲大蠊遺傳進化分析和藥用基因資源挖掘打下了重要基礎[11]。但目前對突變小鼠與正常小鼠之間進行對比分析的基因組測序報道甚少。
因此本實驗對實驗室單獨發現的BALB/c突變卷毛鼠與群體中未發生突變的正常BALB/c小鼠進行測序,并進行了生物信息學分析,結果顯示:正常小鼠與卷毛小鼠產生的Raw Data分別為122.85 Gb和118.04 Gb,過濾后的Clean Data分別為119.82 Gb和112.18 Gb,GC含量分別為40.63%和40.48%,GC分布正常,測序建庫成功。自人類基因組計劃完成以來,獲得高質量的序列圖譜成為了不同物種進行功能基因研究的基礎[12]。而遺傳變異統計結果顯示:卷毛小鼠的雜合分型SNPs總數顯著高于正常小鼠,同時正常小鼠與卷毛小鼠之間累計SNPs深度分布也存在顯著的差異。卷毛小鼠與正常小鼠插入缺失(Indels)以及累計Indels深度分布均存在差異。卷毛小鼠與正常小鼠的SV類型中大片段的插入卷毛小鼠大于正常小鼠,而其他類型則小于正常小鼠。而拷貝數變異(CNVs)結果顯示,正常小鼠和卷毛小鼠的拷貝數增加的個數分別為1 270和967個;而拷貝數減少的個數分別為5 027和3 505個,卷毛小鼠均小于正常小鼠。遺傳變異檢測分析結果顯示SNPs、Indels、SV和CNVs在兩種小鼠之間均存在差異,不僅證明了兩種小鼠存在遺傳差異,且也極大的豐富了遺傳資源多樣性的研究內容[13]。那么在全基因組水平上,利用SNPs等分子遺傳標記存在的差異,也能夠較全面解析物種受到的自然選擇和人工選擇導致的遺傳變化[14]。本實驗對兩種小鼠進行了初步的基因差異對比,但又考慮到即使正常繁育SNP等位點也會發生變異,因此下一步我們將借助一些基因分析軟件[15]對新發現的遺傳變異差異進行詳細的注釋,對差異表達基因的功能進行分類,期待能夠挖掘這些變異對某些分子和細胞事件有所指示。
綜上所述,本研究對BALB/c突變卷毛鼠與同種群中正常BALB/c小鼠進行了全基因組測序,并且發現二者之間存在遺傳變異差異,進一步驗證了前期實驗中兩種小鼠各項指標的差異存在。眾所周知,全基因組測序可以全面、快速、準確地解析不同品種的分子遺傳特征,為品種的不斷選育提高及開發利用奠定堅實的基礎[16]。我們將對其基因的變異情況進行深入分析,以期更好地開發和應用該突變小鼠。
猜你喜歡
興趣閱讀·興趣作文與閱讀(低年級)(2025年8期)2025-08-18 00:00:00
音樂探索(2022年2期)2022-05-30 21:01:37
民用飛機設計與研究(2020年4期)2021-01-21 09:15:02
學苑創造·A版(2020年9期)2020-10-13 09:41:02
小天使·一年級語數英綜合(2019年8期)2019-08-27 02:23:00
電子制作(2018年18期)2018-11-14 01:48:24
小學科學(學生版)(2018年7期)2018-08-13 09:33:04
山東工業技術(2016年15期)2016-12-01 05:31:22
鄭州大學學報(醫學版)(2015年2期)2015-02-27 14:50:46
云南中醫學院學報(2014年3期)2014-07-31 18:57:34
