桂枝甘草湯干預心肌缺血再灌注損傷大鼠心律失常的作用機制
2023-11-21 02:23:46朱明軍唐進法曹英杰王小曉
中西醫結合心腦血管病雜志 2023年21期
關鍵詞:劑量
王 艷,高 原,朱明軍,唐進法,李 彬,王 賀,曹英杰,崔 琳,王小曉,沈 思
心肌缺血再灌注(I/R)損傷是指在急性心肌梗死(acute myocardial infarction,AMI)發生后,藥物或機械的早期再灌注在挽救缺血心肌的同時,會進一步導致心肌細胞功能障礙和組織損傷[1]。其中,心律失常是心肌I/R損傷的重要臨床表現,以室性心律失常最為常見,嚴重者可發生惡性室性心動過速(ventricular tachycardia,VT)和心室纖顫(ventricular fibrillation,VF),是造成病人猝死的重要原因。細胞內鈣超載和心肌能量代謝障礙是誘發心律失常的重要原因,而Na+-K+-ATP酶和Ca2+-Mg2+-ATP酶在維持細胞鈣穩態及能量的產生和利用方面起重要作用[2-4]。心肌I/R發生時Na+-K+-ATP酶和Ca2+-Mg2+-ATP酶活性降低,導致心臟電活動的不穩定性和傳導異常,增加心律失常發生率[5-6]。心肌細胞間信號傳遞的結構基礎是縫隙連接(gap junction,GJ),在心臟電傳導活動中發揮著重要的作用[7-8]。縫隙連接蛋白43(connexin 43,Cx43)則是心肌細胞間縫隙連接的主要連接蛋白,在心肌I/R狀態下其表達含量和自身磷酸化狀態的改變造成心肌細胞間電脫偶聯,導致電沖動傳導異常和心律失常的發生和持續[9-10]。桂枝甘草湯(Guizhi Gancao Decoction,GGD)溫陽益氣,是治療心悸的基礎方,出自漢代張仲景《傷寒論》,原文記載:“發汗過多,其人叉手自冒心,心下悸,欲得按者,桂枝甘草湯主之”。臨床研究表明,GGD對不同類型心律失常具有較好療效[11-12]。現有研究提示,GGD能夠通過減輕鈣超載、清除氧自由基、抑制炎癥反應和細胞凋亡等機制對I/R心肌具有保護作用[13-14]。本研究通過構建大鼠心肌I/R損傷模型,從分子生物學層面研究GGD通過改善Cx43表達對心肌I/R致心律失常的影響及相關機制,為其減輕心律失常的臨床治療提供實驗證據。
1 材料與方法
1.1 動物
Sprague-Dawley(SD)雄性大鼠90只,體質量250~300 g,購自河南省實驗動物中心,動物許可證號為SCXK(豫)2017-0001。
1.2 藥品及試劑
GGD浸膏(按照原著藥量比例:即桂枝、甘草按2∶1的比例制備;1 g浸膏相當于9 g生藥)由河南中醫藥大學第一附屬醫院提供。琥珀酸美托洛爾緩釋片(Met)購自阿斯利康制藥有限公司;血清肌酸激酶同工酶(CK-MB)、肌鈣蛋白I(cTnI)檢測試劑盒(批號:M3U4S6G5BR、78C8ABFAJY)購自Elabscience Biotechnology Co.,Ltd;Na+-K+-ATP酶、Ca2+-Mg2+-ATP酶試劑盒(批號:TF4X7MBJWY、24NWU56XV9)購自Elabscience Biotechnology Co.,Ltd;大鼠Cx43免疫組化試劑盒(批號:15386-1-AP)購自武漢三鷹生物技術有限公司;Kir2.1多克隆抗體(批號:B7301)、磷酸化Cx43(p-Cx43)多克隆抗體(批號:B7101)購自ImmunoWay Biotechnology Company;辣根過氧化物酶(HRP)標記山羊抗鼠二抗(批號:B0101)、HRP標記山羊抗兔二抗(批號:20000258)購自武漢三鷹生物技術有限公司;甘油醛-3-磷酸脫氫酶(GAPDH)抗體(批號:43929)購自GeneTex。
1.3 方法
1.3.1 造模方法
建立大鼠心肌I/R模型[15],末次給藥1 h后,所有大鼠用10%水合氯醛(300 mg/kg,腹腔注射)麻醉后,仰臥位固定,連接PowerLab持續描記心電圖Ⅱ導聯。于頸部正中切開行氣管插管并通過動物呼吸器輔助呼吸,在胸骨左緣第3肋與第4肋間間隙進行開胸手術,暴露心臟,用6-0號帶針縫合線于左前降支(肺動脈圓錐與左心耳交界)處穿線,將一直徑3 mm的乳膠管墊于結扎線與血管之間,系緊縫合線,進行心肌缺血30 min,然后取出墊扎乳膠管,進行再灌注120 min。對照組只穿線不結扎。缺血模型成功標準:結扎部位以下心肌組織變暗、發紺,心電圖ST段明顯抬高。再灌注模型成功標準:缺血區心肌發紺消失、逐漸變紅,抬高的ST段逐漸回落>50%。
1.3.2 分組及給藥方法
將90只大鼠隨機分為對照組、I/R組、GGD低劑量組、GGD高劑量組和美托洛爾組,每組18只。GGD低劑量組、GGD高劑量組分別給予1.8、3.6 g/kg的GGD灌胃,每天1次,連續14 d;美托洛爾組給予美托洛爾9.5 mg/kg灌胃,每天1次,連續14 d;對照組及I/R組給予相同體積生理鹽水。因麻醉或手術失敗導致大鼠意外死亡,各組存活大鼠數量分別為對照組18只,I/R組、GGD低劑量組、美托洛爾組各15只,GGD高劑量組14只。
1.3.3 心電圖指標及評分方法
觀察再灌注后室性期前收縮(ventricualr premature contraction,VPC)、VT或VF的發生率和持續時間,并進行心律失常評分。根據1984年倫敦Lambeth會議確定的評分標準[16]:0分為無心律失常;1分為持續時間≤10 s的VT或其他心律失常,無VF;2分為持續11~30 s的VT或其他心律失常,無VF;3分為持續31~90 s的VT或其他心律失常,無VF;4分為持續91~180 s的VT或其他心律失常,和(或)少于10 s的可逆性VF;5分為持續超過180 s的VT或其他心律失常,和(或)超過10 s的可逆性VF;6分為不可逆性VF。
1.3.4 心肌組織Na+-K+-ATP酶、Ca2+-Mg2+-ATP酶水平的測定
采用分光光度法檢測心肌組織Na+-K+-ATP酶、Ca2+-Mg2+-ATP酶的含量。制備心肌組織勻漿,用考馬斯亮藍和光譜法測定蛋白含量,比色定磷法測定,Na+-K+-ATP酶、Ca2+-Mg2+-ATP酶按照試劑盒的說明書進行測定。
1.3.5 血清CK-MB、cTnI水平測定
采用酶聯免疫吸附試驗(ELISA)法測定血清CK-MB、cTnI水平含量。再灌注120 min后腹主動脈取血,靜置30 min后,以2 000 r/min離心15 min(取上清液)。按照ELISA試劑盒步驟測定血清中CK-MB、cTnI含量。
1.3.6 心肌組織的病理學觀察
HE染色法觀察心肌組織病理形態學改變。再灌注120 min后取出心臟,用冷的磷酸緩沖鹽溶液(PBS)沖洗。取心肌組織轉移至4%甲醛中浸泡,乙醇脫水,石蠟包埋切片(5 μm),用蘇木精-伊紅(HE)染色,在200倍放大鏡下觀察心肌組織的變化。
1.3.7 心肌組織Cx43表達的測定
采用免疫組化法檢測心肌組織Cx43表達。取心肌組織轉移至4%甲醛中浸泡,乙醇脫水,石蠟包埋切片(5 μm),脫蠟和抗原修復后,加3%的H2O2阻斷內源性過氧化物酶,加一抗(1∶100),加酶標二抗,進行二氨基聯苯胺(DAB)顯色,蘇木精復染,脫水和封片,鏡檢拍照顯微鏡下觀察。
1.3.8 心肌組織中p-Cx43和Kir2.1的蛋白表達
采用蛋白免疫印跡(Western Blot)法測定心肌組織中p-Cx43和Kir2.1的蛋白表達。提取心肌組織總蛋白,聚丙烯酰胺凝膠電泳,轉移至聚偏二氟乙烯膜,用脫脂奶粉封閉后,分別加p-Cx43抗體(1∶1 000)和Kir2.1抗體(1∶1 000),與GAPDH一抗(1∶1 000)平行進行孵育過夜,TBST沖洗,加HRP標記羊抗兔免疫球蛋白G(IgG)或HRP標記的山羊抗鼠IgG為二抗(1∶2 000),孵育1 h,應用化學發光試劑顯色,使用Image J圖像分析軟件分析各蛋白條帶的吸光度,蛋白表達水平以目的蛋白條帶IOD值/GAPDH的IOD值表示。
1.4 統計學處理
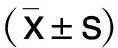
2 結 果
2.1 GGD對心肌I/R大鼠心律失常的影響
用PowerLab連續監測心電圖(見圖1),分析再灌注期間心電圖的變化,監測再灌注后心律失常發生率、持續時間和心律失常評分。結果顯示,對照組大鼠在心電監測期間偶發VPC、手術時引起的VT,但無VF發生;其他組大鼠均發生VPC、VT或VF,且以VT及VF為主。與對照組相比,I/R組VPC次數明顯增多,VT及VF持續時間明顯延長,且心律失常評分明顯增高(P<0.01),表明造模成功;與I/R組比較,GGD高劑量組、美托洛爾組VPC的發生次數明顯減少,VT及VF的持續時間明顯縮短,心律失常評分明顯降低,差異均有統計學意義(P<0.05)。詳見表1。
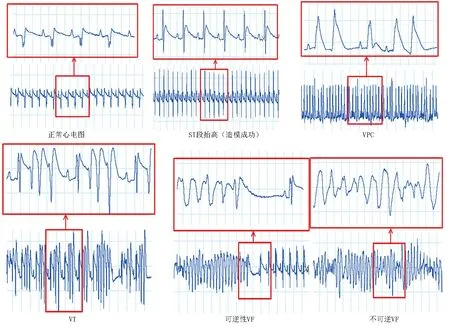
圖1 PowerLab連續監測心電圖示例
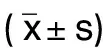
表1 各組VPC發生次數、心律失常持續時間、心律失常評分比較
2.2 GGD對心肌I/R損傷大鼠血清CK-MB、cTnI水平的影響
與對照組相比,I/R組大鼠CK-MB、cTnI水平明顯升高(P<0.01);與I/R組相比,GGD低劑量組、GGD高劑量組和美托洛爾組CK-MB、cTnI水平明顯降低(P<0.01)。詳見圖2、圖3。
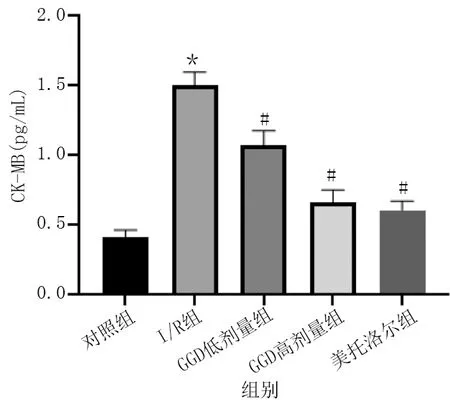
圖2 各組血清CK-MB水平比較
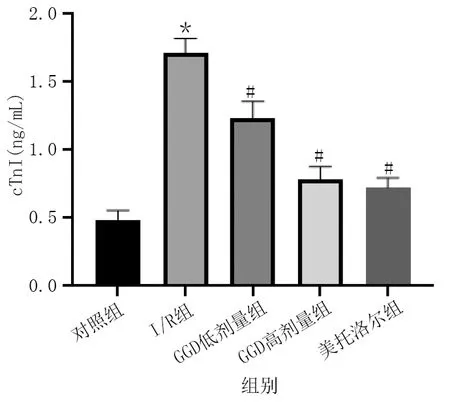
圖3 各組血清cTnI水平比較
2.3 GGD對心肌I/R損傷大鼠心肌組織中Na+-K+-ATP酶和Ca2+-Mg2+-ATP酶水平影響
與對照組相比,I/R組大鼠Na+-K+-ATP酶和Ca2+-Mg2+-ATP酶活性明顯降低(P<0.01);與I/R組相比,GGD低劑量組、GGD高劑量組和美托洛爾組Na+-K+-ATP酶和Ca2+-Mg2+-ATP酶活性明顯升高(P<0.05或P<0.01)。詳見圖4、圖5。
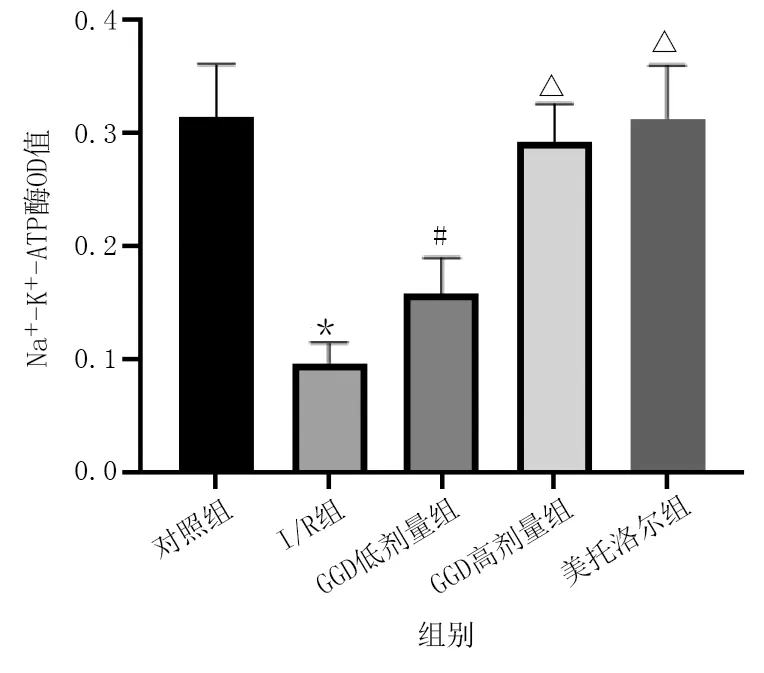
圖4 各組心肌組織Na+-K+-ATP酶活性比較
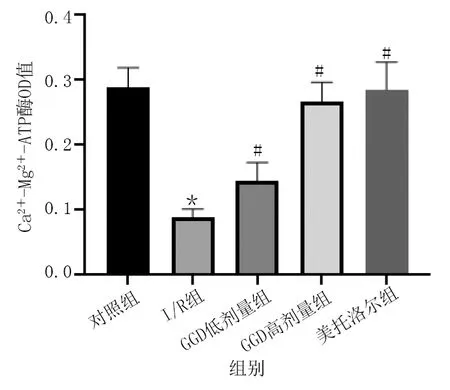
圖5 各組心肌組織Ca2+-Mg2+-ATP酶活性比較
2.4 GGD對心肌I/R損傷大鼠心肌組織作用的病理學改變
對照組心肌細胞結構清楚,心肌纖維細胞整齊,未見明顯異常;I/R組心肌細胞排列紊亂,著色不均,心肌纖維腫脹、斷裂;與I/R組比較,GGD低劑量組、GGD高劑量組和美托洛爾組心肌形態相對整齊,且隨GGD劑量增加,心肌細胞內水腫和炎性細胞浸潤明顯減輕。詳見圖6。
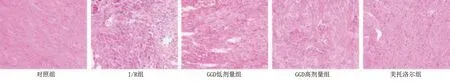
圖6 各組大鼠心肌組織病理學變化(HE染色,×200)
2.5 GGD對心肌I/R損傷大鼠Cx43、p-Cx43和Kir2.1蛋白表達的影響
免疫組化結果顯示:對照組可見較多棕黃色Cx43蛋白表達,分布規律,呈條帶狀;I/R組Cx43表達明顯降低,呈點狀無規律分布;與I/R組相比,GGD低劑量組、GGD高劑量組和美托洛爾組Cx43表達明顯增多,分布較規律。詳見圖7。Western Blot結果顯示:與對照組比較,I/R組p-Cx43和Kir2.1蛋白表達下降(P<0.01);與I/R組比較,GGD高劑量組和美托洛爾組p-Cx43和Kir2.1蛋白表達升高(P<0.05或P<0.01)。詳見圖8。

圖7 免疫組化檢測各組Cx43蛋白表達(×400)
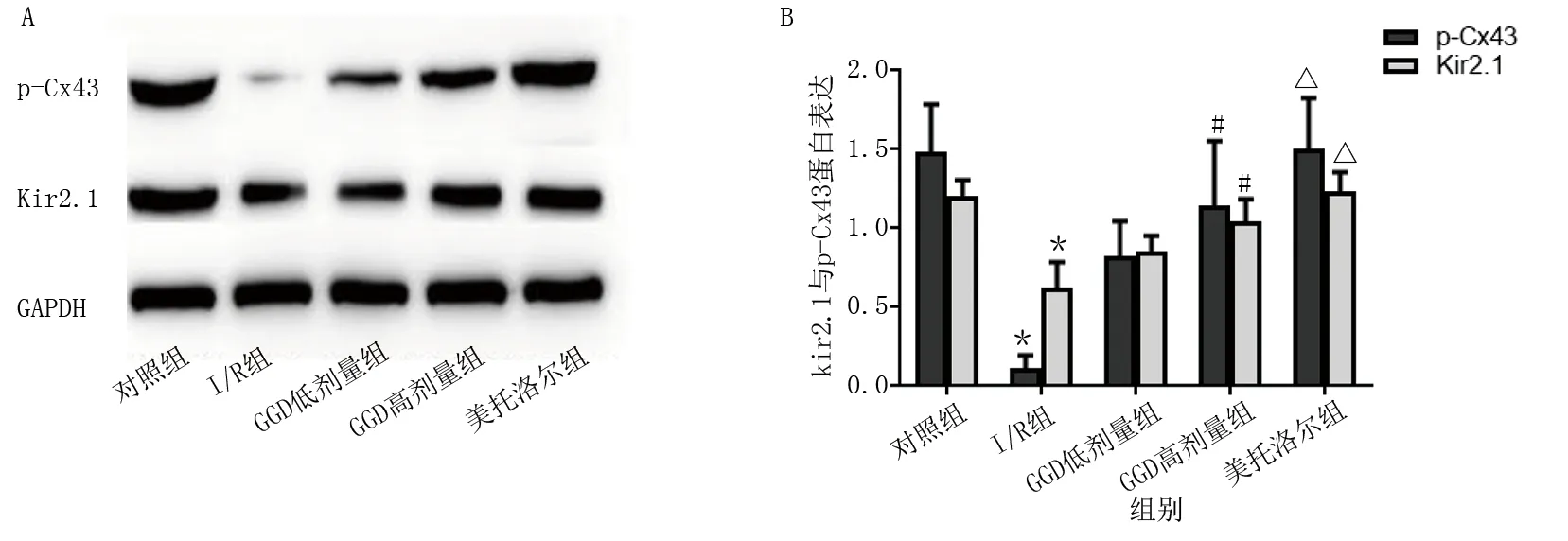
圖8 Western Blot檢測各組心肌組織中p-Cx43和Kir2.1蛋白水平
3 討 論
GGD是張仲景治療心悸的基礎方,臨床報道其具有很好的抗心律失常作用[11-12]。基礎研究發現,GGD不僅能改善多種實驗性心律失常[14,17],還可縮小心肌I/R大鼠心肌梗死面積從而發揮心肌保護作用,其機制涉及減輕鈣超載、抗氧化、抑制炎癥反應和細胞凋亡,調控TLR4/NF-κB信號轉導通路等多個方面[13,18]。李冀等[19]發現桂枝甘草湯提取物30醇組分、水組分能夠抗氯化鋇、烏頭堿及哇巴因等誘發的心律失常,機制可能與減少心肌細胞L型Ca2+、Na+通道電流及延長心肌細胞動作電位時程(APD)和抑制動作電位幅度(APA)有關。GGD乙酸乙酯部位的30%乙醇洗脫組分與肉桂酸協同對小鼠心律失常有一定的保護作用[20]。GGD及各組分含藥血清均有抑制鈣離子通道作用,該作用與鈣離子阻滯劑相同[18]。本研究通過建立大鼠心肌I/R模型,探討GGD對Cx43蛋白表達、Na+-K+-ATP酶活性、Ca2+-Mg2+-ATP酶活性、Kir2.1蛋白表達的影響。
心肌梗死病人發生心肌壞死、細胞膜通透性增加,使心肌細胞中CK-MB、cTnI等心肌損傷標志物大量釋放入血,其升高水平與心肌細胞損傷程度關系密切[21-22]。本研究結果顯示,I/R組大鼠血清CK-MB和cTnI水平較對照組明顯升高,GGD藥物組血清CK-MB和cTnI水平明顯降低,HE染色結果也進一步表明GGD藥物組較對照組可減輕心肌損傷程度,提示GGD藥物組可改善大鼠心肌I/R損傷。
心肌I/R致心律失常的發生機制主要包括氧自由基堆積、鈣超載、能量代謝障礙等幾個方面。Na+-K+-ATP酶又稱鈉泵,其功能為水解ATP酶產生能量逆電化學梯度跨膜轉運Na+和K+,維持細胞內外Na+、K+離子濃度梯度,Ca2+-Mg2+-ATP酶即為鈣泵,可跨細胞膜主動將Ca2+轉運到細胞外并主動攝取Ca2+從胞質中進入肌漿網。Na+-K+-ATP酶和Ca2+-Mg2+-ATP酶在維持細胞內鈣穩態方面發揮著重要作用。當心肌I/R損傷發生時,心肌細胞能量代謝障礙,ATP生產減少,Na+-K+-ATP酶活力下降,細胞內Na+增多,K+減少,激活Na+/Ca2+交換使胞內Ca2+濃度增高,同時,Ca2+-Mg2+-ATP酶活性也下降,不能將胞內過多的Ca2+排出或攝入肌漿網,導致鈣超載,誘發心律失常[23-25]。本研究結果表明,GGD藥物組Na+-K+-ATP酶和Ca2+-Mg2+-ATP酶活性明顯升高,有利于維持細胞內鈣穩態,改善心肌細胞能量代謝,從而達到拮抗心肌缺血再灌注心律失常的作用。
縫隙連接通道分布于心肌細胞閏盤處,主要由3種連接蛋白構成,包括Cx40、Cx43、Cx45,其中心室肌的連接蛋白以Cx43為主,其正常表達及分布是保證心肌細胞間電耦聯活動和協調心肌舒縮功能的關鍵因素[26]。生理狀態下,Cx43有磷酸化和去磷酸化(Np-Cx43),但僅p-Cx43構成有功能的縫隙連接通道[27]。在心肌I/R損傷發生時,Cx43蛋白將表現脫磷酸化,間隙連接處的結構和功能變化,導致心肌組織阻力增加,導電性降低;間隙連接處的電性斷開,導致電沖動的傳導變慢,電活動合并,最終導致嚴重的室性心律失常。縫隙連接處的結構和功能變化,導致心肌組織阻力增加、電導性降低,出現縫隙連接的電脫耦連現象,導致電沖動傳導變慢和折返性電活動的發生,最終導致嚴重的室性心律失常[28-29]。IK1的主要組成為Kir2.1,由KCNJ2基因編碼,在維持細胞靜息電位及鉀離子電位平衡調節中發揮重要作用,是構成心肌細胞動作電位復極末期的主要電流通道之一[27,30]。在心肌缺血再灌注期間,心肌組織Kir2.1蛋白表達下調引起IK1下降,導致靜息膜電位去極化,延長了動作電位時程,增加了心律失常風險[31-32]。本研究結果表明,I/R組Cx43蛋白的分布明顯降低,p-Cx43和Kir2.1蛋白表達水平下降,GGD高劑量組、低劑量組Cx43表達明顯增多,并在一定程度上逆轉了p-Cx43和Kir2.1蛋白表達的下調,這可能是GGD改善心肌I/R心律失常的相關機制。
綜上所述,GGD可減少VPC的發生次數,縮短VT及VF的持續時間,改善心律失常評分,其減輕心律失常的相關機制可能與改善Cx43的表達和磷酸化狀態、提高Na+-K+-ATP酶和Ca2+-Mg2+-ATP酶活性及上調Kir2.1蛋白表達水平有關。通過提高ATP酶的活性,改善細胞能量代謝,維持心肌細胞內鈣穩態,恢復細胞間縫隙連接蛋白功能與IK1電流,從而縮短動作電位時程與復極化進程,降低激動傳導速度不均一,消除或減少折返電活動的發生,達到抑制心律失常的作用。
猜你喜歡
課堂內外·初中版(科學少年)(2023年10期)2023-12-10 00:43:06
全科護理(2022年10期)2022-12-26 21:19:15
中國合理用藥探索(2022年1期)2022-11-26 00:22:32
今日農業(2022年4期)2022-11-16 19:42:02
鄉村科技(2021年33期)2021-03-16 02:26:54
國際放射醫學核醫學雜志(2021年10期)2021-02-28 08:41:58
藥學與臨床研究(2015年4期)2015-06-05 11:35:54
衛生職業教育(2014年24期)2014-05-20 09:05:38
同位素(2014年2期)2014-04-16 04:57:20
中國合理用藥探索(2014年11期)2014-03-11 20:30:20
