
正交試驗優化消乳增生軟膏的提取工藝研究
2023-12-07 09:03:08皮鳳娟張慶蓮張盛敏
中國民族民間醫藥 2023年22期
關鍵詞:實驗
羅 川 皮鳳娟 張慶蓮 許 曾 張盛敏
四川省瀘州市中醫醫院藥劑科,四川 瀘州 510500
乳腺增生為常見乳腺疾病,又稱“乳腺結構不良”“纖維囊性乳腺病”,屬病理性增生,主要表現為正常乳腺小葉的生理性增生與復舊不全及乳腺正常結構出現紊亂[1]。其主要臨床表現為乳房腫塊和脹痛,是一種既非炎癥又非腫瘤的增生性病變[2],其發病率占乳腺疾病的首位[3]。消乳增生軟膏為我院協定處方,原方為散劑,用水調勻后直接外敷于痛處,起到軟堅散結,活血止痛的效果。本方用藥精煉,但各中藥成分藥效有所差異,因此在對本復方的提取工藝研究中不能只以單味藥材的提取工藝作為參照,而應考慮本復方的整體性。本文根據中藥復方的整體性,在保留傳統中藥散劑特點的基礎上[4],分別以有效成分陳皮和芥子中的橙皮苷[5]、芥子硫氰酸鹽含量及干浸膏得率為指標,采用單因素實驗法和正交實驗法對其制備工藝進行研究,為以后開發成院內制劑奠定基礎。
1 儀器與材料
1.1儀器 FA2004萬分之一電子天平(上海安亭電子儀器廠),GZX-GF101-3BS-Ⅱ/H電熱恒溫鼓風干燥箱(上海躍進醫療器械廠),HH數顯恒溫水浴鍋(上海躍進醫療器械廠),CQ-250A-DST超聲清洗儀(上海躍進醫用光學器械廠),ZDHW調溫電熱套(北京中興偉業儀器有限公司),1260高效液相色譜儀(安捷倫科技有限公司)。
1.2 材料 芥子硫氰酸鹽對照品(批號111702-202107,含量以100%計)、橙皮苷對照品(批號110721-202019,含量以95.3%計)均購自中國食品藥品檢定研究院;陳皮(批號210700791)、芥子(批號190700581)、肉桂(批號210701091)等均由成都市康美藥業生產有限公司提供,山奈(批號200924-1)和穿破石(批號190910-1)由瀘州百草堂中藥飲片有限公司提供,胡椒(批號20070103)由四川國強中藥飲片有限公司提供。上述試藥經檢驗均為真品,所使用的試劑等級:乙腈為色譜純,其余試劑為分析純。
2 方法與結果
2.1 單因素考察
2.1.1 吸水率考察 按照處方量稱取藥材共96 g,加入1000 mL水浸泡,分別在10 min、20 min、30 min、60 min、120 min、180 min測定濾液體積。吸水率=(1000-濾液體積)/1000。測定該處方吸水率為11%。
2.1.2 加水量考察 按照處方量的兩倍準確稱量藥材(丁香、陳皮、肉桂等)3份,固定浸泡時間為30 min,提取次數為1次,加水量依次為6倍、8倍、10倍,提取7 h,并記錄每小時揮發油的體積,揮發油總體積分別為3.4 mL、2.9 mL、2.5 mL。結果表明:在加水量為6倍時所提取到的揮發油較多。故選擇加水量為6倍。結果如圖1所示。

圖1 加水量對揮發油提取體積的影響圖
2.1.3 浸泡時間考察 準確稱量兩倍處方量的藥材(肉桂、陳皮、丁香等)3份,固定提取次數為一次,加水量為6倍[6],浸泡時間分別為1 h[7]、1.5 h,提取7 h,并記錄每小時揮發油的體積,揮發油總體積分別為3 mL、2.8 mL。綜合2.1.1浸泡30 min的結果,表明:在浸泡時間為30 min時所得揮發油較多,故選擇浸泡時間為30 min。結果如圖2所示。

圖2 浸泡時間對揮發油提取體積的影響圖
2.1.4 提取時間考察 結合2.1.1和2.1.2兩個實驗,在提取時間超過5 h后,揮發油體積增長緩慢,考慮到生產成本等因素,故選擇最佳提取時間為5 h。結果如圖3所示。

圖3 提取時間對揮發油提取體積的影響圖
2.2 含量測定
2.2.1 色譜條件 色譜柱:ZORBAX-SB-C18(250 mm×4.6 mm,5 μm)。流動相:1 mL/L乙腈∶含0.1%磷酸的0.08 mol/L磷酸二氫鉀溶液(20 ∶80)。柱溫:30 ℃。檢測波長0~7 min ∶326 nm。7~15 min ∶283 nm;流速:1 mL/ min[8-10]。進樣量10 μL。

A.橙皮苷對照品溶液;B.芥子硫氰酸鹽對照品溶液;C.供試品溶液;D.缺芥子陰性對照品溶液;E.缺陳皮陰性對照品溶液圖4 專屬性考察高效液相色譜圖
2.2.2 對照品溶液的制備 芥子硫氰酸鹽對照品溶液的制備:取芥子硫氰酸鹽對照品適量,精密稱定,加不含0.1%磷酸的流動相制成每1 mL含0.22 mg的對照品貯備液[11],精密量取對照品貯備液2 mL,置10 mL容量瓶中,用不含0.1%磷酸的流動相稀釋并定容,即得;橙皮苷對照品溶液的制備:取適量橙皮苷對照品,精密稱定,加甲醇制成每1 mL含0.52 mg的對照品貯備液,精密量取對照品貯備液1 mL,置10 mL容量瓶中,用甲醇稀釋并定容,即得。
芥子陰性對照品溶液的制備:除去芥子以外的其他藥材,按優選提取工藝制備陰性樣品,再按供試品溶液制備方法制備陰性對照品溶液。陳皮陰性對照品溶液的制備:取除去陳皮以外的其他藥材,按優選提取工藝制備陰性樣品,再按供試品溶液制備方法制備陰性對照品溶液。
2.2.3 供試品溶液的制備 精密量取正交實驗項下制備的溶液5 mL,共9份,置于具塞錐形瓶中,水浴蒸干后加入甲醇10 mL,精密稱定,超聲 45 min,放冷,再次稱定重量,用甲醇補足損失的重量,搖勻,濾過,取續濾液即得[11]。
2.2.4 方法學考察
2.2.4.1 專屬性實驗 分別精密量取2.2.2項下的芥子硫氰酸鹽對照品溶液、橙皮苷對照品溶液、缺芥子陰性對照品溶液、缺陳皮陰性對照品溶液和2.2.3項下的供試品溶液,各10 μL,注入高效液相色譜儀,按上述色譜條件檢測。結果對照品溶液和供試品溶液在相同的保留時間均有出峰,陰性對照無干擾,如圖4。說明該測定方法專屬性強,無干擾。
2.2.4.2 線性范圍考察 精密量取2.2.2項下的芥子硫氰酸鹽對照品貯備液和橙皮苷對照品貯備液各1 mL、2 mL、3 mL、4 mL、5 mL、6 mL、7 mL、8 mL,分別置于10 mL容量瓶中,芥子硫氰酸鹽對照品貯備液用不含0.1%磷酸的流動相,橙皮苷照品貯備液用甲醇稀釋并定容,按2.4.1項下的色譜條件進樣測定,以進樣量(X,μg)為橫坐標,得回歸方程分別為:y1=614.14x-49.4,r=0.9999(n=8),y2=974.1x-28.711,r=0.9999(n=8)。結果表明:芥子硫氰酸鹽在0.22~1.76 μg內,橙皮苷在0.52~4.16 μg范圍內,進樣量與峰面積呈良好的線性關系。
2.2.4.3 精密度實驗 精密量取2.2.2項下芥子硫氰酸鹽對照品溶液和橙皮苷對照品溶液,分別重復測定6次,結果芥子硫氰酸鹽、橙皮苷峰面積的RSD值分別為0.69%,0.72%,表明該方法精密度良好。
2.2.4.4 穩定性實驗 精密量取2.2.2項下芥子硫氰酸鹽對照品溶液和橙皮苷對照品溶液,分別在0 h、2 h、4 h、6 h和8 h分別進樣10 μL,記錄峰面積,計算RSD分別為0.56%,0.78%,結果表明芥子硫氰酸鹽和橙皮苷對照品均在8 h內穩定。
2.2.4.5 重復性實驗 取同一批樣品6份,照2.2.3項下方法制備供試品溶液,按照2.2.1項下色譜條件進行測定,記錄峰面積,計算含量。結果芥子硫氰酸鹽對照品溶液和橙皮苷對照品溶液的RSD值分別1.2%和1.1%,表明本方法的重復性良好。
2.2.4.6 加樣回收率實驗 精密量取已知芥子硫氰酸鹽和橙皮苷含量的樣品2.5 mL,分別取6份,置于具塞錐形瓶中,每份均精密加入濃度為0.5180 mg/mL橙皮苷對照品儲備液5 mL,濃度為0.2208 mg/mL芥子硫氰酸鹽對照品儲備液5 mL,照2.2.3項下方法制備供試品溶液,依照上述色譜條件平行操作測定,記錄峰面積,計算回收率,結果RSD<3.0%,說明加樣回收率好。
2.2.5 品的含量測定 分別精密吸取2.2.2項下制備的對照品溶液和2.2.3項下制備的供試品溶液各10 μL,按2.2.1項下的色譜條件進樣測定,計算樣品中橙皮苷和芥子硫氰酸鹽的含量。
2.3 干浸膏得率的測定 精密量取正交項下制備的提取液每個25 mL,置于已干燥至恒重的蒸發皿中,水浴蒸干,再置于105 ℃烘箱中恒重,計算干浸膏得率[12]。
2.4 正交實驗 考慮本次試驗是采用水做溶劑,因此選擇加水量、提取時間和提取次數作為正交實驗的考察因素,將干浸膏得率和藥液中的芥子硫氰酸鹽含量、橙皮苷含量作為考察指標進行綜合評定,應用L9(34)正交實驗表進行實驗,因素與水平表見下表1。按照L9(34)正交實驗設計要求,準確稱取處方量藥材 (陳皮、丁香、肉桂等),共9份,編為1~9號。 先按照單因素試驗的最佳提取條件,進行揮發油提取,將提取好揮發油的藥材棄去藥液,留下藥渣待用,向上述藥渣中加入處方中余下藥材,按照表1因素加水進行提取,將提取液過濾、冷卻和定容。結果見表2。

表1 提取工藝的因素與水平

表2 正交實驗設計與結果
2.5 多指標評分方法建立 實驗以芥子硫氰酸鹽和橙皮苷的含量及干浸膏得率作為權重系數依據,將芥子硫氰酸鹽和橙皮苷的含量權重系數分別設置為0.3,將干浸膏得率的權重系數設為0.4。綜合評分=(各組芥子硫氰酸鹽含量/芥子硫氰酸鹽最高含量×0.3×100)+(各組橙皮苷含量/橙皮苷最高含量×0.3×100)+(各組干浸膏得率/干浸膏最高得率×0.4×100)。
2.6 方差分析結果 由方差結果分析可知,各因素影響芥子中芥子硫氰酸鹽及陳皮中橙皮苷提取量的排序B>C>A。結果表明,加水量A對橙皮苷和芥子硫氰酸鹽的含量和干浸膏得率影響不顯著 (P>0.05),但提取次數B和提取時間C對橙皮苷和芥子硫氰酸鹽的含量和干浸膏得率影響均具有顯著性 (P<0.01)。因此確定最佳提取工藝條件為A1B2C1,即加6倍量水,煎煮2次,每次15 min。結果見表3。

表3 方差分析結果
2.7 驗證實驗 由于正交實驗中篩選的最佳條件為6倍加水量,提取兩次,每次15 min,做1組驗證實驗。考慮到本次試驗提取時間短,且提取次數無顯著性影響,因此在考察提取時間1 h和1.5 h,從驗證結果來看,提取時間為1.5 h時含量較高,1 h次之,15 min含量最少,但提取1.5 h的比提取1 h的時間成本高,且多出的有效成分有限,為減少資源浪費,最終選擇的最佳提取工藝條件為:加水量6倍,提取2次,每次1 h。結果見表4。
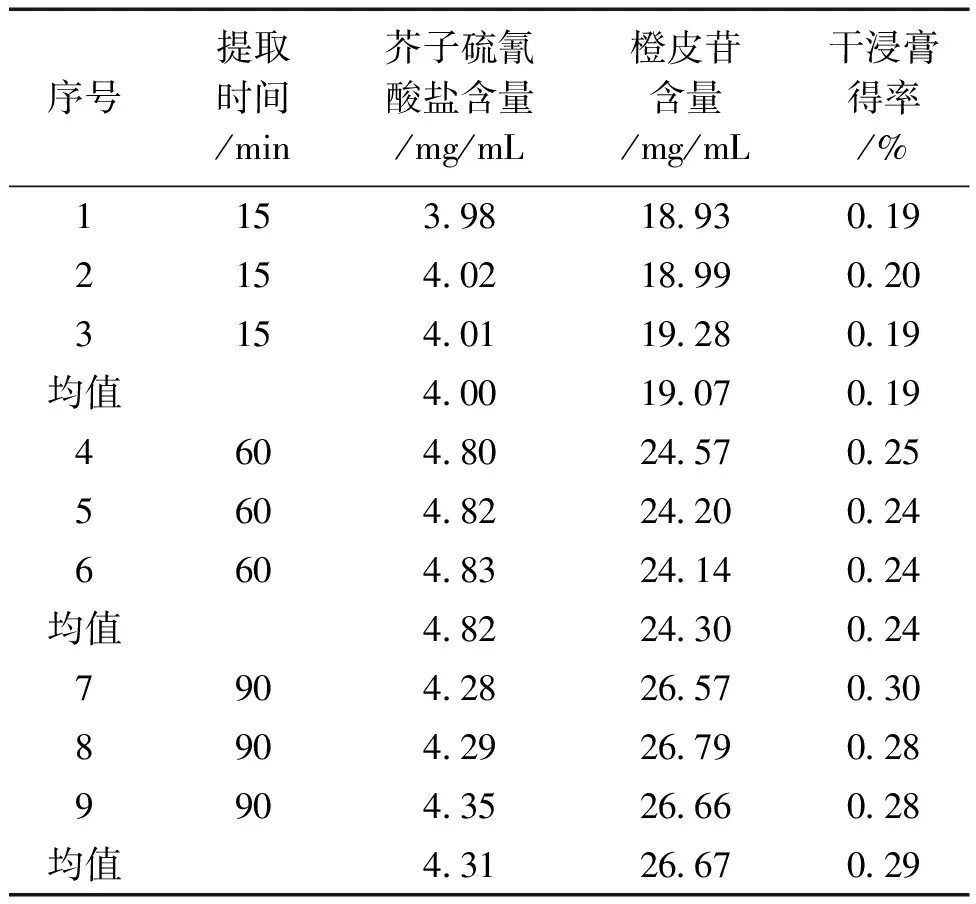
表4 驗證實驗結果
3 討論
在此次提取工藝研究實驗中,因需考察兩個有效成分的含量,故選用合適的高效液相色譜條件比較重要,最初采用的流動相條件為乙腈:0.08 mol/L 磷酸二氫鉀溶液(10∶90),在此條件下芥子硫氰酸鹽對照品的保留時間長,且存在拖尾現象。后將流動相比例調整為20∶80,保留時間合適,但也存在拖尾現象,因此向流動相中加入0.3%的三乙胺溶液,拖尾現象也未解決。查閱資料后,因芥子硫氰酸鹽顯酸性,故向流動相中加入0.1%的磷酸調整PH,拖尾現象得以解決。也曾考察不同的色譜柱[ZORBAX-SB-C18柱,Speax Bio-C18柱,Eclipse XDB-C18柱,Speax MS-C18柱],對峰型和分離度的影響,結果顯示ZORBAX-SB-C18柱的峰型和分離度最好。研究所確定的色譜條件,可為后續含芥子和陳皮的實驗研究提供參考。
從單因素結果來看,當加水量在6倍時,所得的揮發油含量較高,表明在相同時間內,6倍加水量的揮發油提取效率最高。此外,在浸泡時間考察中,浸泡30 min即達到揮發油提取的最大值。提取時間在4~6 h之間呈現平穩趨勢,在6h時達到峰值,但在提取時間超過5 h后,揮發油體積增長緩慢,考慮到生產成本等因素,故選擇最佳提取時間為5 h。由上述結果可以看出,該處方中所含的揮發油易于提取,適合大生產。
消乳增生軟膏為我院協定處方,具有軟堅散結,活血止痛的效果,原方為散劑,存在劑量不準確,難保存且患者依從性低的缺點,本實驗通過采用單因素和正交試驗相結合的方式對消乳增生軟膏的提取工藝進行研究,確定了消乳增生軟膏的最佳提取工藝,提高了有效成分含量,從驗證結果來看,提取時間為1.5 h時含量較高,1 h次之,15 min含量最少,但提取1.5 h的比提取1 h的時間成本高,且多出的有效成分有限,為減少資源浪費,最終選擇的最佳提取工藝條件為:加水量6倍,提取2次,每次1 h。但本次試驗提取方法單一,下一步打算驗證其不同提取方法下有效成分的含量變化。
猜你喜歡
作文·小學低年級(2025年2期)2025-02-13 00:00:00
小雪花·小學生快樂作文(2024年11期)2024-12-31 00:00:00
作文·小學低年級(2024年2期)2024-04-29 00:00:00
作文·小學低年級(2023年3期)2023-04-29 00:00:00
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
小主人報(2022年4期)2022-08-09 08:52:06
中學生數理化·中考版(2022年11期)2022-02-16 07:01:20
小哥白尼(趣味科學)(2019年6期)2019-10-10 01:01:50
發明與創新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
