注射用頭孢美唑鈉的質量分析
2024-01-01 04:31:18耿悅劉文欣宋蕓峰龐慶林張錦琳侯玉榮袁耀佐曹玲張玫
中國抗生素雜志 2023年8期
耿悅 劉文欣 宋蕓峰 龐慶林 張錦琳 侯玉榮 袁耀佐 曹玲 張玫
摘要:目的 評價注射用頭孢美唑鈉的質量現狀。方法 采用調研、文獻檢索、法定標準檢驗結合探索性研究的方式,對國內20家企業生產的177批次的注射用頭孢美唑鈉及1批原研制劑的質量進行比較分析;通過對雜質譜、成鹽率、配伍穩定性、包材相容性、無菌保障水平等關鍵質量屬性進行考察,分析不同企業產品的質量差異。結果 通過法定標準檢驗,177批次注射用頭孢美唑鈉均符合規定,但仍然存在部分項目檢驗方法和限度不統一,個別藥品說明書不完善等問題。探索性研究中,采用LC/MS對雜質結構進行推斷,對其來源進行歸屬。優化了有關物質檢查方法。配伍穩定性結果表明,個別說明書需要修訂。在無菌保障水平方面,國內企業在微生物的鑒定溯源能力方面還有待進一步加強。結論 ? ?注射用頭孢美唑鈉符合現行標準要求,現行標準有待統一和提高。
關鍵詞:注射用頭孢美唑鈉;評價性抽驗;質量分析;雜質譜;成鹽率;配伍穩定性;包材相容性;無菌保障水平
中圖分類號:R978.1 ?文獻標志碼:A
Quality analysis of cefmetazole sodium for injection
Geng Yue1,2, Liu Wenxin1,2, Song Yunfeng3, Pang Qinglin1, Zhang Jinlin1,2, Hou Yurong1,2,
Yuan Yaozuo1,2, Cao Ling1,2, and Zhang Mei1,2
(1 Jiangsu Institute of Food and Drug Control, Nanjing 210019; 2 NMPA Key Laboratory for Impurity Profile of Chemical Drugs,
Nanjing 210019; 3 Nanjing University of Traditional Chinese Medicine, Nanjing 210029)
Abstract ? ?Objective ? ?To evaluate the quality status of cefmetazole sodium for injection. Methods ? ?177 batches of cefmetazole sodium for injection produced by 20 enterprises and one batch of the original drug were compared and analyzed by means of investigation, literature search, the legal standard test, and exploratory research. By investigating the key quality attributes such as impurity spectrum, salt formation rate, packaging material compatibility, and sterility guarantee level, the quality differences of products from different enterprises were analyzed. Results ? ?Statutory tests showed that 177 batches of cefmetazole sodium for injection all met the requirements. But there were still some problems, such as inconsistent inspection methods and limits of some items, imperfect individual drug instructions, and so on. Through exploratory research, the impurity structures were inferred by LC/MS and their sources were attributed. The inspection methods of relevant substances were optimized. Compatibility stability results indicated that individual instructions needed to be revised. In terms of sterility assurance level, the identification and traceability ability of domestic enterprises needed to be further strengthened. Conclusion ? ?The overall quality of cefmetazole sodium for injection met the requirement of the current statutory standards, but the current standard needed to be unified and improved.
Key words Cefmetazole sodium for injection; Evaluative random test; Quality analysis; Impurity spectrum; Salt Formation rate; Compatible stability; Compatibility of packaging materials; Sterility guarantee level
頭孢美唑鈉是日本第一三共株式會社研制的第二代頭孢菌素類抗生素。頭孢美唑鈉具有廣譜、高效、低毒的作用,對革蘭陽性和陰性菌均有抗菌作用[1]。與其他頭孢菌素類抗菌藥物相比,由于其頭孢烯母核7位a-H被甲氧基取代,增加了母核的立體阻礙,提高了對β-內酰胺酶的穩定性,故對青霉素類和頭孢菌素類抗菌藥物的耐藥菌株仍有很強的抗菌活性[2]。臨床上注射用頭孢美唑鈉主要用于治療敗血癥、急性支氣管炎、肺炎等,不良反應主要包括皮疹、瘙癢癥以及過敏性休克等[3]。
中國藥典(ChP)、美國藥典(USP)、日本藥典(JP)和韓國藥典(KP)[4-7]均收載了頭孢美唑鈉原料藥。ChP2020、JP18和KP10收載了注射用頭孢美唑鈉, USP43收載了注射用頭孢美唑。經查詢國家藥品監督管理局(NMPA)進口藥品和國產藥品批準文號數據庫,國內上市的注射用頭孢美唑鈉規格分別為0.5、1.0、1.5和2.0 g。本次評價性抽驗樣品涉及13個檢驗標準,為藥典標準(經標準比對,ChP2015和ChP2020注射用頭孢美唑鈉的檢驗方法和限度均一致,以下簡稱ChP2020/2015)以及12個國家食品藥品監督管理局標準。
為加強對上市后藥品質量的監管,NMPA分別在2013年[8]和2021年將注射用頭孢美唑鈉列入年度國家藥品評價性抽驗計劃。評價抽驗的目的在于客觀地評價國內上市藥品的質量現狀;分析產品的主要質量問題進而明確提高產品質量的方向。本研究在完成2021年評價性抽驗法定標準檢驗的基礎上,進一步開展探索性研究,結合法定檢驗與探索性研究結果,對國內上市注射用頭孢美唑鈉的質量狀況進行評價,并從安全性、有效性的角度進行客觀評價并提出建議。本次國家藥品評價性抽驗從全國28個省、自治區和直轄市的生產企業、經營單位以及使用單位抽取到177批樣品,樣品渠道來源占比分別為32.2%、52.5%和15.3%,其對應的原料涉及5家生產企業。注射用頭孢美唑鈉為頭孢美唑的無菌粉末,不含任何輔料,故本研究同時可反映頭孢美唑原料藥的質量狀況。
1 儀器與試藥
1.1 儀器
Shimadzu LC-20AB高效液相色譜儀、Agilent 1290 Infinity高效液相色譜儀、DionexICS-5000+離子色譜儀,Agilent 6546 Q-TOF質譜儀、Agilent 6470LC/TQ三重四極桿質譜儀、Agilent ICP-MS 8900電感耦合等離子體質譜儀、ADMET Predictor軟件(Version 7.2.0001)、Millipore去離子水發生裝置、梅特勒托利多XS205電子天平。
1.2 試藥
177批次注射用頭孢美唑鈉均為2021年度國家藥品評價性抽驗樣品,涉及20個生產企業(用A至T企業表示),購買到1批次原研制劑(日本第一三共,批號:JPA1110),頭孢美唑對照品(中國食品藥品檢定研究院,批號:130580-202002,純度99.4%),1-甲基-5-巰基-四氮唑(中國食品藥品檢定研究院,批號:510048-201401,純度97.2%),甲醇(色譜級和質譜級,Merck)、乙腈(色譜級和質譜級,Merck)、乙酸銨(質譜級,Sigma)、冰乙酸(質譜級,阿拉丁)、甲酸(質譜級,阿拉丁)、甲磺酸(分析純,Sigma-Aldrich)、磷酸(分析純,國藥集團化學試劑有限公司)、四丁基氫氧化銨(分析純,麥克林)、超純水(Millipore儀制備)。
2 試驗方法
2.1 法定標準檢驗
分別按照各企業現行標準對177批注射用頭孢美唑鈉進行檢驗。主要檢驗項目包括鑒別、含量測定、有關物質及頭孢美唑聚合物等。
2.2 探索性研究
2.2.1 雜質譜的研究
采用多中心切割2D-LC-HRMS法,Agilent 1290 Infinity UPLC與6546 Q-TOF質譜儀聯用分析技術,一維色譜條件同ChP2020/2015有關物質項下,二維色譜條件采用Acquity UPLC BEHC8(1.7 μm, 2.1 mm×
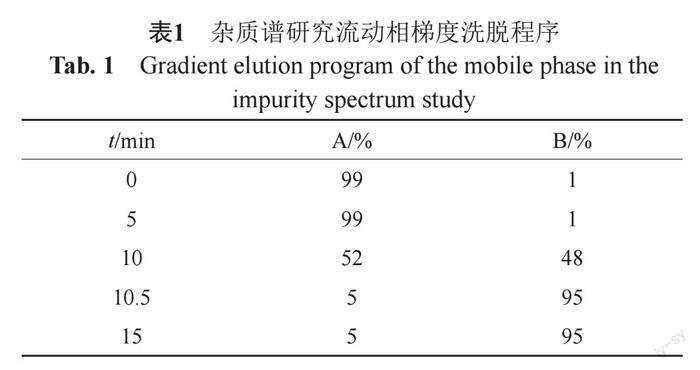
100 mm)色譜柱,流動相為含有1%甲酸的醋酸銨(5 mmol/L)水溶液-1%甲酸乙腈溶液,梯度洗脫(表1)。質譜檢測器采用ESI源,正離子掃描,鞘氣溫度:320℃,鞘氣流速11.0 L/min,噴嘴電壓:1 kV,毛細管電壓:3.5 kV,霧化氣壓力:35 psig,干燥氣溫度:320℃,干燥氣流速:8.0 L/min,毛細管出口氣壓:115 V,碰撞能量:10、20和40 eV,全掃描范圍m/z 100~1700。
2.2.2 有關物質方法的優化
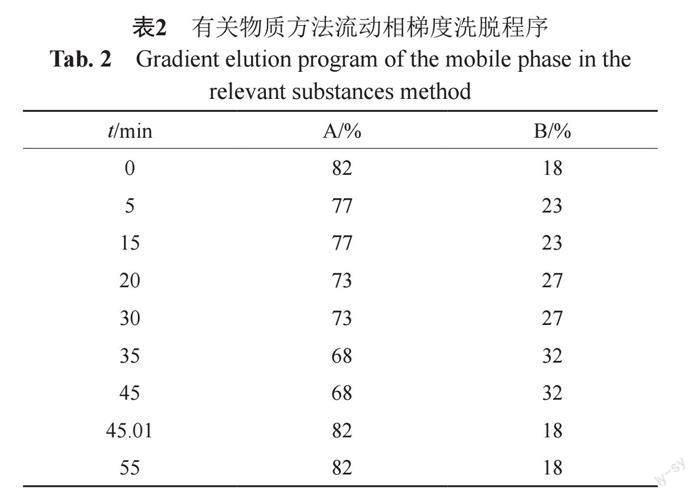
采用資生堂OSAKA SODA CAPCELL PAK MGⅡC18色譜柱(4.6 mm×250 mm, ?5 μm),流動相為流動相A為磷酸二氫銨溶液(取磷酸二氫銨5. 75 g,加水730 mL 使溶解,加10%四丁基氫氧化銨溶液9.6 mL,加四氫呋喃6 mL)(用磷酸調節pH值至4.5),流動相B為甲醇溶液(取甲醇220 mL,加10%四丁基氫氧化銨溶液9.6 mL,加四氫呋喃6 mL),梯度洗脫,見表2,柱溫為35℃;流速為1.0 mL/min;檢測波長為254 nm;進樣體積20 μL。
2.2.3 聚合物雜質的研究
采用高效液相色譜-串聯四極桿飛行時間質譜(HPLC-Q-TOF-MS)檢測技術。色譜條件使用XBridge Shield RP 18(4.6 mm×250 mm,5 μm),流動相A為10 mmol醋酸銨溶液(用醋酸調節pH至4.5),流動相B為甲醇,梯度洗脫,流速為0.8 mL/min,柱后三通分流,檢測波長為254 nm,柱溫為35 ℃。質譜檢測器采用ESI源,正離子模式掃描,鞘氣溫度:320 ℃,鞘氣流速11.0 L/min,噴嘴電壓:1 kV,毛細管電壓:3.5 kV,霧化氣壓力:35 psig,干燥氣溫度:320 ℃,干燥氣流速:8.0 L/min,毛細管出口氣壓:115 V,Skimmer電壓:65 V,八極桿電壓:750 V;碰撞能量:10、20和40 eV,全掃描范圍m/z 100~1700。
2.2.4 成鹽率的考察
采用IonPac CS12A陽離子交換色譜柱(4 mm× 250 mm),IonPac CG12A保護柱(4 mm×50 mm),流動相為4 mmol/L甲磺酸水溶液。抑制電導檢測器,抑制電流設置為59 mA;非抑制電導法采用普通電導檢測器。
2.2.5 配伍穩定性研究
模擬臨床使用方法,將樣品分別用5%葡萄糖注射液、0.9%氯化鈉注射液和注射用蒸餾水溶解稀釋制成1 mg/mL的溶液,分別于0、3、6、9、12、15、18、21和24 h取配伍溶液適量,按ChP2020/2015有關物質檢查法進行測定。
2.2.6 包材相容性研究
(1) 注射劑瓶玻璃元素遷移試驗
采用Agilent 8900 ICP-MS Triple Quad電感耦合等離子體質譜儀進行測定,測定模式為He模式,等離子模式為高基體HMI模式,RF功率1550 W,采樣深度10.0 mm,等離子體流量15 L/min,霧化器流量0.67 L/min,氦模式氦氣流量4.5 mL/min,測定方法為內標法,內標溶液濃度為Bi、Ge、In 500 μg/L, Sc 1000 μg/L,元素7Li、11B、27Al、30Si、47Ti、51V、52Cr、55Mn、56Fe以45Sc為內標,元素59Co、60Ni、63Cu、66Zn、75As、78Se以72Ge為內標,元素95Mo、101Ru、103Rh、105Pd、107Ag、114Cd、118Sn、121Sb、138Ba以115In為內標,元素189Os、193Ir、195Pt、197Au、201Hg、205TI、206Pb以209Bi為內標。
(2) 膠塞中13種亞硝胺遷移檢測
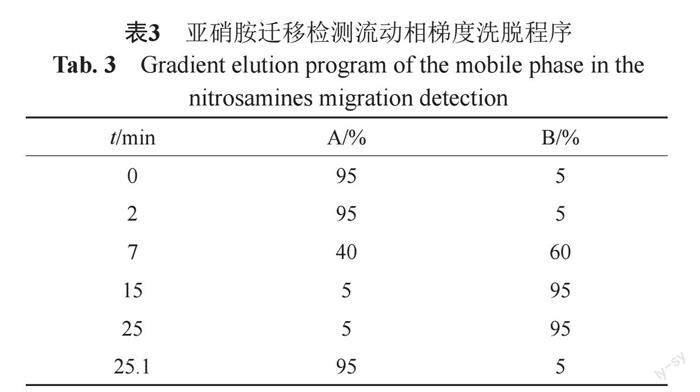
采用Agilent 1290超高效液相與Agilent 6470LC/TQ三重四極桿質譜聯用技術,色譜條件采用ACQUITY UPLC BEH C8(2.1 mm×100 mm, ?1.7 μm)色譜柱和Agilent Infinity Lab Poroshell120 Bonus-RP(3.0 mm×100 mm, ? ? ? 2.7μm)色譜柱分別檢測8個(NDELA、NMOR、NMPhA、NEPhA、NDiBA、NDBA、NDBzA、NPYR)和5個(NDMA、NDEA、NPIP、NDiPA、NDPA)亞硝胺類雜質,A相:0.1%甲酸-水溶液,B相:甲醇,梯度洗脫,見表3。質譜檢測器采用APCI源,正離子掃描,掃描方式:MRM,霧化器溫度:300℃,霧化器壓力:45 psi,毛細管電壓:3000 V。
2.2.7 無菌保障情況的調研
經查閱NMPA 2020年5月14日發布的《化學藥品注射劑仿制藥質量和療效一致性評價技術要求》、藥品生產質量管理規范(2010年修訂)附錄1無菌藥品、美國藥典USP<1229>無菌保障章節、FDA 無菌藥品生產指南(2004版)、EN GMP附錄1無菌藥品生產(2020版),結合該品種無菌生產工藝,對原輔料與包材、設施設備、清潔消毒、環境監控、人員/無菌操作及超標/偏差調查這6部分內容進行探索性研究,形成注射用頭孢美唑鈉無菌保障調查問卷。向本次抽驗涉及的20個企業發放調查問卷,并對反饋的調查問卷進行匯總研究。
3 試驗結果
3.1 法定標準檢驗的結果
按現行質量標準檢驗,177批注射用頭孢美唑鈉全部符合規定。
3.2 法定標準檢驗發現的問題
雖然按法定標準檢驗所有樣品均合格,但有2批樣品中有關物質檢查項結果為邊緣產品:其他次大單雜達上限(0.5%);另部分企業裝量差異偏大。
本品種執行標準有13個,有關物質檢測方法有8種,且雜質命名混亂。現行中國藥典有關物質方法無法將本次檢測涉及的所有雜質有效分離,方法中只規定了“1-甲基-5-巰基四氮唑”“單雜”和“總雜”的限度,未對相關特定雜質的結構進行明確并制訂相應的限度。
3.3 探索性研究結果
根據法定檢驗中發現的問題,結合注射劑一致性評價的相關要求,分別對雜質譜,有關物質、頭孢美唑聚合物雜質、成鹽率、包材相容性進行了研究,由于無法對各企業的無菌保障工藝進行實地考察,因此通過發放問卷的方式調查各企業的無菌保障水平并進行調研。
3.3.1 雜質譜的研究
ChP2020/2015采用HPLC法測定有關物質,JP17和KP10采用薄層色譜法測定,USP43有關物質項缺失。ChP2020/2015中注射用頭孢美唑鈉有關物質的測定方法在專屬性、靈敏度方面遠遠優于其他各部藥典方法。在法定檢驗中,100余批次樣品采用ChP2020/2015有關物質的測定方法檢出的相關雜質達20個以上,但僅控制1個特定雜質(1-甲基-5-巰基四氮唑),其余雜質作為單個雜質含量和雜質總量規定限度,課題組對藥典方法檢出雜質進行了定性分析。
ChP2020/2015有關物質測定方法的流動相含有較高濃度的四丁基氫氧化銨和磷酸二氫銨,與質譜檢測的要求不匹配,若重新建立適合LC/MS檢測的色譜系統,雖可對檢出雜質的結構進行推定,但卻無法實現對藥典條件下色譜圖中各雜質峰的準確指認。因此本實驗采用多中心2D-LC-HRMS法,在不改變原流動相條件的基礎上,實現在線分離和檢測。基于多中心切割在線除鹽-2D-LC-HRMS分析技術,一維色譜系統為藥典系統,將藥典系統的主要雜質一個個收集在loop環中,二維色譜系統將目標物洗脫進入二維色譜柱,對目標物完成快速脫鹽,然后進行質譜分析。對12個含量大于0.05%的雜質進行了結構推測,根據出峰時間編號1~12(圖1)。與已知雜質的保留時間和質譜數據比較,歸屬了4個色譜峰為已有對照品的雜質。另外,基于頭孢美唑和已知雜質的色譜行為和質譜信息以及前期匯總的頭孢美唑鈉雜質信息[9-10],對另外8個雜質進行了結構推測(12個主要雜質的質譜信息及歸屬見表4,雜質結構信息見圖2)。通過強制降解實驗,對主要雜質的來源進行分析,其中9個雜質為降解雜質(限于篇幅,本文僅將研究結果進行描述,具體研究過程另文發表)。
3.3.2 有關物質方法的優化
β-內酰胺類抗生素由于其自身結構的不穩定以及生產工藝的特點,產品中的有機雜質種類復雜、含量相對較高,因此分析控制其有機雜質對優化生產過程、提高臨床用藥安全性至關重要。本次抽驗品種,有關物質有8種檢測方法,限度要求也不一致,提示本品有關物質檢查項應統一修訂提高。所有有關物質檢測方法均為等度洗脫,等度洗脫存在洗脫及分離能力較弱的問題,不利于疏水性較強的雜質的洗脫,且雜質命名混亂。雜質譜研究結果顯示,現行中國藥典有關物質方法,無法將本次檢測涉及的所有雜質有效分離,并未對相關特定雜質的結構進行明確并制訂相應的限度。為了更好了解不同廠家樣品的雜質分布情況,并對其進行統一的評價,本文通過對梯度洗脫程序等因素的考察,對中國藥典有關物質檢測方法進行了改進(“2.2.2”項),增強了對雜質的洗脫能力及相互間的有效分離,并對關鍵雜質進行了結構確證和命名,見圖2。
為更好地對國評產品的雜質檢出情況進行比較,在探索性研究中首先采用藥典有關物質方法對法定檢驗未采用藥典方法檢測的樣品以及一批原研樣品進行了檢測。在完成有關物質方法優化之后,對本次國評征集到的20家生產企業樣品隨機抽取1批,及一批原研制劑進行了有關物質探索性研究檢測,與原方法相比,各雜質之間分離更佳,尤其在雜質較多的樣品中可以分離出更多雜質(圖3)。研究發現,28家生產企業的樣品(來自5家原料企業)雜質譜基本一致,僅雜質量有差異。結合調研中企業提供的穩定性數據,確定了2個指針性雜質。新建立有關物質方法中雜質1是產品中的最大單個雜質(雜質譜研究中確認為頭孢美唑內酯),也是最重要的降解雜質,反映制劑的儲存情況;雜質12(雜質譜研究中確認為頭孢美唑MMT異構體)是大部分產品的次大單個雜質,其既是工藝雜質又是降解雜質,反應了原料的合成工藝水平及儲存情況。日本原研產品所含雜質種類、分布、含量與國產注射用頭孢美唑鈉相比,均較一致,提示國產制劑與日本市售原研藥的雜質水平相當。
3.3.3 頭孢美唑聚合物雜質的研究
文獻報道[11],頭孢美唑鈉因其結構特點(頭孢美唑結構式見圖4),側鏈上沒有活性基團氨基,且7位側鏈上含有甲氧基取代,位阻較大,聚合途徑比較單一,聚合物的含量非常低,各種破壞均難以生成聚合物雜質。法定檢驗中:175批樣品采用G10法測得的頭孢美唑聚合物量在0.001%~0.01%之間,小于制劑中雜質0.05%的報告閾值;2批樣品采用TSK法對聚合物進行控制。諸多文獻的研究結果表明,利用分子排阻色譜法檢測β-內酰胺類抗生素的聚合物雜質存在著“專屬性較差”的問題,也就是可能出現“保留時間小于主成分的雜質也并不全是高分子雜質”的情況[12-14]。筆者將TSK法頭孢美唑主峰之前雜質峰切換到二維液相色譜系統進行質譜分析,亦暫未發現聚合物雜質。
隨著對β-內酰胺類抗生素的聚合反應機制、各種聚合物結構及聚合物與其他降解雜質的關系等的研究不斷深入,利用“指針性雜質”-HPLC能檢出的寡聚物雜質來間接控制高分子聚合物雜質的策略應用得越來越廣泛。本文建立了反相色譜系統常規質譜方法(“2.2.3”項),測定到一個可能的頭孢美唑聚合物雜質(圖5)。該雜質(tR=18.235 min)在一級質譜圖中的準分子離子峰[M+H]+、[M+NH4]+的分別為m/z 827.0817、844.1084推測其分子式為C28H30N10O10S5,在二級質譜圖中出現了豐度較高的m/z 472、m/z 356以及m/z 215的頭孢美唑特征碎片離子,結合頭孢美唑鈉聚合物研究資料,我們將該雜質歸屬為頭孢美唑聚合物。通過其EIC圖和色譜圖,我們可以發現該雜質的含量非常低,遠低于制劑中雜質0.05%的報告閾值。在UV圖譜中很難對聚合物峰進行確認,所以將聚合物雜質作為特定雜質,在反相色譜系統進行控制的難度較大。
G10法費時耗力,目前研究表明TSK法主峰之前未發現聚合物雜質。是否需要對頭孢美唑聚合物單獨控制還有待進一步商榷。從檢驗經濟學角度出發,建議刪節藥典標準中聚合物測定方法,可作為企業內控項目。
3.3.4 成鹽率的考察
成鹽率是藥品非常關鍵的質量屬性,成鹽率影響藥物的溶解度、酸堿度以及溶液的顏色等。若頭孢美唑酸成鹽不完全,剩余的游離頭孢美唑酸過多,會引起藥物血漿蛋白結合率高,起效時間延長。USP43、JP17和KP10均規定了含量測定以無水物計結果的上限,頭孢美唑酸若成鹽不完全,含量會偏高。
本文首先在文獻報道[15-16]的基礎上,優化建立了離子色譜抑制電導檢測器測定鈉離子的方法,方法專屬性好,靈敏度高,但離子色譜價格昂貴,對實驗人員要求較高,儀器普及率較低,課題組又進一步建立非抑制電導法測定鈉離子的含量。非抑制電導法,只需要給普通高效液相色譜儀配置一個電導檢測器,就可以滿足基層檢測的需要,操作簡單。非抑制電導法雖然靈敏度略低于離子色譜法,但專屬性好、準確性高。兩種方法均對本次國評征集到的20家生產企業樣品隨機抽取1批,及一批原研制劑進行了鈉離子的測定。同時按照企業各自的法定檢驗標準測定頭孢美唑的含量。成鹽比%=(鈉離子含量/22.99)/(頭孢美唑含量/471.54),22.99和471.54分別為鈉離子和頭孢美唑的摩爾質量。供試品中鈉與頭孢美唑的比值,理論值為1(1:1)。各企業產品成鹽比無明顯差異,國內產品與進口產品二者成鹽均較完全,成鹽比在0.99~1.04。
3.3.5 配伍穩定性試驗
仔細研讀各國產企業注射用頭孢美唑鈉的藥品說明書,發現用法用量中均有注明:靜脈注射時,取本品溶于注射用蒸餾水,生理鹽水或葡萄糖注射液。一家企業的藥品說明書中寫道溶解后應盡快使用,室溫下保存不超過24 h。藥品說明書與臨床使用情況息息相關。取該企業的樣品開展配伍穩定性試驗,該企業生產的注射用頭孢美唑鈉樣品與5%葡萄糖注射液、0.9%氯化鈉注射液和注射用蒸餾水配伍,均不穩定。1-甲基-5-巰基四氮唑的含量顯著增加,24 h內,在葡萄糖溶液中由0.42%增加至0.89%,在0.9%氯化鈉溶液中由0.51%增加至0.99%;一個未知雜質(新建有關物質方法中的雜質5)含量在3種配伍溶液中均顯著增加,由0.02%左右增至0.5%左右;其他最大單雜不穩定(雜質譜研究中確認為頭孢美唑內酯),其含量隨時間變化而顯著降低;所以總雜質的量呈現先減少后增加的趨勢。主成分的含量也有降低。建議該企業修訂藥品說明書為臨用新制。
3.3.6 包材相容性研究
本次抽驗樣品注射劑是一種起效快、高風險的劑型,其包材相容性一直廣受關注,抽驗樣品的內包裝均為玻璃注射劑瓶和膠塞。對本次國評征集到的20家生產企業樣品隨機抽取1批,及一批原研制劑進行了元素雜質和亞硝胺類雜質的遷移試驗。遷移試驗測定了樣品中所有來源元素雜質和亞硝胺類雜質的總量。
經ICP-MS/MS法考察砷、鎘、汞、鉛、鈷、鎳、釩、金、銥、鋨、鈀、鉑、銠、釕、銀、硒、鉈、鋇、鉬、鉻、銅、鋰、銻、錫、硼、鋁、硅、鈦、錳、鐵、鋅等31種元素的遷移情況,結果顯示,國產制劑和原研制劑,元素雜質向藥粉中的遷移量無明顯升高,且均在ICH-Q3D規定的[17]元素雜質每日最大允許攝入量(PDE)的30%以內。
采用UPLC-MS法測定國產與原研制劑中13種亞硝胺類物質的含量。結果顯示,10個國產企業樣品中有N-亞硝基哌啶(NPIP)檢出,但均低于定量限(最低定量濃度為0.024 μg/g),其他亞硝胺類雜質均未檢出,原研產品未檢出亞硝胺類雜質(最低檢出濃度為0.012 μg/g)。國家藥監局藥審中心發布的《化學藥物中亞硝胺類雜質研究技術指導原則(試行)》指出,長期暴露于高出可接受水平的致突變性致癌物如NDMA可能會增加患癌風險,但持續70年每天服用含有等于或低于可接受水平NDMA的藥品的人并不會增加患癌風險。注射用頭孢美唑鈉并不是終生長期治療用(>10年)藥物,且NPIP檢出量均低于定量限,根據測定結果看,目前注射用頭孢美唑鈉中亞硝胺類物質的風險可控。
3.3.7 無菌保障情況的調研
設計調研問卷,對本次抽驗涉及的20家企業在原料與包材、設施設備、清潔消毒、環境監控、人員/無菌操作及超標/偏差調查等方面進行調研,全部收到反饋,調研結果表明:①該品種不涉及輔料,為原料直接分裝;生產工藝主要包括鋁蓋/膠塞/注射劑瓶的清洗滅菌干燥,原料藥外包裝的清潔消毒,分裝加塞,軋蓋,包裝等環節。核心灌裝區的加工環境為常規潔凈室B+A,或限制進入隔離系統(RABS)或隔離器系統;②生產過程中嚴格控制原料的無菌和細菌內毒素、包裝材料的微生物負載和細菌內毒素;在環境檢測與微生物控制方面,通過對人員的培訓考核、生產環境中生產中對懸浮粒子和微生物監測、設施設備的清潔消毒等方式控制生產的環境,保證了產品的無菌水平;③對非物料接觸表面或間接接觸表面的清潔消毒控制中,85%的企業在消毒劑的使用之前進行了消毒劑效果評價;80%企業對進入潔凈區物品的清潔消毒程序中考慮了不同物品的特性,并分別制定清潔消毒措施;20家企業在消毒中均使用了殺孢子劑。通過對消毒劑質量的控制及清潔消毒效果的驗證,保證了生產的環境和設備的潔凈度,保證了產品的無菌水平;④對于產品中檢出微生物及生產中微生物的鑒定溯源能力有待進一步加強,在調研的20家企業中70%的企業會對日常環境監控分離微生物進行鑒定,60%的企業定期對環境分離微生物的鑒定結果進行總結,并進行群落分析。微生物鑒定技術還有待進一步加強,10%的企業僅采用顯微鏡進行形態分析;45%的企業采用手工生化試劑條或半自動/自動生化鑒定設備進行生化水平的分析;20%的企業可采用基因測序等分子生物學技術進行基因水平的分析。對微生物鑒定中僅60%的企業能達到種的水平。
數據分析結果顯示,本品國內企業的無菌控制水平均可滿足相關指導原則要求,但在微生物的鑒定溯源能力方面還有待進一步加強。
4 結論
本次國家評價性抽驗采用前期調研、文獻檢索、法定標準檢驗結合探索性研究的方式對注射用頭孢美唑鈉的質量現狀進行評價和分析。按照法定標準檢驗,177批次樣品均符合規定,說明注射用頭孢美唑鈉質量總體評價較好,但有2批次抽驗樣品其他次大單雜達上限,部分企業裝量差異偏大,需要引起重視。質量標準方面,本品種執行標準有藥典標準及12個國家食品藥品監督管理局標準,有關物質檢測方法有8種,且雜質命名混亂;頭孢美唑聚合物測定方法費時耗力,且檢出雜質遠低于制劑中雜質0.05%的報告閾值,質量標準有待統一及提高。
通過探索性研究,先后采用多中心切割2D-LC-HRMS系統及LC-Q-TOF不含鹽系統,分析注射用頭孢美唑鈉雜質結構,研究其雜質譜及來源,通過對不同來源樣品的雜質譜進行分析,發現國產注射用頭孢美唑鈉與原研產品的雜質譜基本一致,提示國內企業與原研企業的頭孢美唑合成工藝水平基本相當;建立了有關物質梯度方法,建議有關物質項下增加對特定雜質的控制,本品為原料藥直接灌裝的制劑,建議同時開展原料的質量標準提高工作;配伍穩定性研究表明,一家企業應盡快修訂藥品使用說明書;無菌保障水平方面,國內企業的無菌控制水平均可滿足相關指導原則要求,但在微生物的鑒定溯源能力方面還有待進一步加強。
參 考 文 獻
于守汎. 頭孢美唑[J]. 國外醫藥抗生素分冊, 1995, 16(4): 259-264.
陸紅彬, 胡延維, 楊磊. 頭孢美唑合成工藝的研究進展[J].抗感染藥學, 2014, 11(1): 6-12.
盧玨. 26例頭孢美唑致患者不良反應發生的影響因素分析[J]. 抗感染藥學, 2019, 16(9): 1555-1558.
中國國家藥典委員會. 中華人民共和國藥典: 2020年版二部[S]. 北京: 中國醫藥科技出版社, 2020: 340-341.
The United States Pharmacopeial Convention, The United States Pharmacopeial43-The National Formulary38[S]. (Volume III). Rockville, 2020: 824.
Pharmaceutical and Medical Device Regulatoryscience Society of Japan, Japanese Pharmacopeia XVII[S]. Tokyo, ?2016: 627-628.
Ministry of Food and Drug Safety. Korean Pharmacopoeia 10: part1[S]. Cheongwon County, 2017: 252-253.
楊志強, 劉釤, 鄭挺, 等. 國產注射用頭孢美唑鈉的質量評價[J]. 海峽藥學, 2021, 33(9): 38-42.
鄒文博, 陳振賀, 王明娟, 等. 采用二維高效色譜-串聯四級桿飛行時間質譜法對注射用頭孢美唑鈉的未知雜質進行結構解析[J]. 中國藥學雜志, 2022, 57(8): 645-650.
陳秀明, 鄭孝賢, 周璟明, 等. UPLC-Q-TOF-MS分析頭孢美唑鈉中有關物質[J]. 中國抗生素雜志, 2020, 45(10): 1017-1026.
胡昌勤. β-內酰胺抗生素聚合物分析技術的展望[J]. 中國新藥雜志, 2008, 17(24): 2098-2102.
裘亞, 秦峰, 聞宏亮, 等. 國產注射用氯唑西林鈉的質量分析與研究[J]. 中國抗生素雜志, 2016, 41(9): 666-670.
秦峰, 丁穎, 裘亞, 等. 在線二維凝膠色譜反相液相色譜飛行時間質譜技術對鹽酸頭孢替安中聚合物雜質的分析研究[J]. 中國藥學雜志, 2016, 51(9): 742-750.
李進, 張培培, 姚尚辰, 等. 注射用哌拉西林鈉他唑巴坦鈉的聚合物雜質分析[J]. 藥物分析雜志, 2019, 39(7): 1279-1294.
錢敏, 耿志旺, 彭茗, 等. 離子色譜法測定頭孢曲松鈉中的鈉離子含量以及成鹽率[J]. 藥物分析雜志, 2015, 35(3): 435-439.
譚勝連, 閔翠娥, 李毅, 等. HPLC-CDD法測定以碳酸鈉為助溶劑的頭孢類產品中碳酸鈉的含量[J]. 中國抗生素雜志, 2012, 37(12): 920-923.
人用藥品注冊技術要求國際協調會. ICH指導原則Q3D元素雜質指導原則[EB/OL]. (2020-12-25) [2021-07-14].https://www.ich.org/page/guality-guidelines#3-6.
