無定形Fe基MOF材料的制備及析氧性能研究
2024-01-03 11:18:58秦王昕上官向陽王康軍于廣莉
沈陽化工大學學報 2023年3期
關鍵詞:催化劑
秦王昕, 上官向陽, 王康軍, 于廣莉
(沈陽化工大學 化學工程學院, 遼寧 沈陽 110142)
隨著經濟與科學技術的快速發展,能源危機和環境惡化問題日益嚴峻,因此清潔能源得到了廣泛的需求和推廣[1-2].在眾多可替代能源中,氫能(H2)因來源廣、能量密度高等優點被認為是一種有前途的可再生能源[3],并且H2是一種零碳排的能源載體,水是唯一燃燒產物,因而開發利用H2是實現2050年碳中和目標的有效途徑.電解水是生產高純H2的有效方法之一,但陽極析氧反應(OER)涉及復雜4e-轉移過程,需要較大的過電位才能克服動力學遲緩問題,從而限制了整個電解水制氫的效率[4-5].目前,RuO2、IrO2等貴金屬氧化物對OER具有優異的電催化性能,但其固有的金屬稀缺性、高昂的成本阻礙了其大規模應用.因此,開發廉價、高效的OER催化劑仍然是一項巨大挑戰.
晶態金屬有機框架(MOF)是由無機節點(金屬離子或簇)與有機配體通過配位鍵自組裝而形成的一類新型多孔材料.因其高度有序的多孔性、大的比表面積和可調節的金屬活性中心等優勢,MOF在氣體儲存/分離、催化、藥物緩釋和傳感等方面具有潛在的應用前景[6-12].近年來,具有過渡金屬中心的MOF材料(如CoFe-MOF、FeNi-MOF和NiCo-MOF等)在電催化領域取得了初步進展[13-15].然而,MOF良好的結晶度是一把雙刃劍,多數活性位點被有機配體包裹而難以得到有效利用,大大降低了其催化活性.與晶態MOF材料相比,無定形MOF(aMOF)保留了晶態MOF的基本結構單元和連通性,缺乏長程有序的網格結構,已逐漸引起科研工作者的關注.aMOF材料不僅具有較高的熱、化學穩定性,而且金屬中心的配位數較低,大量的缺陷促使材料表面以及內部暴露出更多的活性位點.迄今為止,aMOF材料已初步應用于藥物緩釋和氣體吸附等研究領域[16-18],在電催化方面的研究較少.
筆者采用外來金屬離子誘導策略,以均苯三甲酸為有機配體制備了一系列無定形的InxFey-aMOF材料.研究發現:Fe3+擾亂了In3+與有機配體配位,致使原始晶態In-MOF失去長程有序結構而暴露出更多的本征活性位點;Fe-aMOF表現出最佳的電催化性能,電流密度為0.01 A/cm2時的過電位僅為258.2 mV,塔菲爾斜率為45.4 mV/dec,優于大多數傳統的晶態MOF材料;同時該催化劑還具有優異的長期穩定性.
1 實驗部分
1.1 實驗試劑
九水合硝酸鐵[Fe(NO3)3·9H2O,分析純]、水合硝酸銦[In(NO3)3·xH2O,質量分數為99%]、1,3,5-均苯三甲酸(H3BTC,質量分數為98%)和氫氧化鉀(KOH,質量分數為99%),上海阿拉丁生化科技股份有限公司;導電碳黑(VXC-72R),卡博特有限公司;Nafion溶液(質量分數為5%),上海兢翀電子科技發展有限公司;泡沫鎳(NF,厚1.0 mm),昆山廣嘉源新材料有限公司;無水乙醇(C2H5OH,分析純)、鹽酸(HCl,質量分數為36%~38%)、丙酮(C3H6O,分析純)和N,N-二甲基甲酰胺(DMF,分析純),國藥集團化學試劑有限公司.
1.2 InxFey-aMOF的制備
In1Fe1-aMOF的制備過程如下:首先,將450 mg(1.50 mmol)Fe(NO3)3·9H2O和604 mg(1.50 mmol)In(NO3)3·xH2O先后溶解于10 mL DMF和40 mL水的混合溶液中,攪拌15 min后,加入660 mg(3.14 mmol)1,3,5-均苯三甲酸,繼續攪拌20 min形成均一混合液;其次,將上述混合液轉移至100 mL高壓反應釜中,并在120 ℃烘箱中晶化5 d;再其次,混合液冷卻至室溫,用離心方法(10 000 r/min,10 min)收集固體粉末,依次用DMF、水、乙醇將粉末洗滌3次,除去未反應雜質;最后,將得到的產物在100 ℃真空烘箱中干燥8 h,命名為In1Fe1-aMOF.
金屬離子總物質的量保持為3.0 mmol,通過改變Fe(NO3)3·9H2O和In(NO3)3·xH2O初始物質的量的比來調控產物中金屬的組分,即In、Fe的初始物質的量的比為1∶0、1∶3、1∶4和0∶1,相應產物分別命名為In-MOF、In1Fe3-aMOF、In1Fe4-aMOF和Fe-aMOF,其制備方法與In1Fe1-aMOF相同.
1.3 實驗儀器
采用Rigaku SmartLab X射線衍射儀表征電催化劑的晶體結構(λ=0.154 18 nm,40 kV,30 mA),掃描范圍為4°~40°,掃描速率為10 (°)/min;采用Nicolet iS50傅里葉變換紅外光譜儀對電催化劑的官能團進行測試,測試范圍為400~4 000 cm-1;采用蔡司ZEISS Gemini 300掃描電子顯微鏡觀察催化劑的形貌、尺寸和元素分布;采用FEI TECNAI G2F20透射電子顯微鏡分析催化劑的結構、有序性等微觀信息;采用康塔Autosorb-IQ-C全自動物理吸附儀表征催化劑的比表面積和孔結構,測試前樣品在120 ℃下真空活化10 h;采用Thermo Fisher iCAP 7400電感耦合等離子體原子發射光譜儀對催化劑的組成進行定量測試.所有催化劑的電化學性質均在上海辰華CHI 600E型電化學工作站上進行.
1.4 電化學性能測試
1.4.1 泡沫鎳的預處理
首先,將商用泡沫鎳(NF)剪成1 cm × 2 cm,在丙酮中超聲清洗3次,去除表面油污;其次,在1 mol/L鹽酸溶液中超聲清洗3次,去除表面氧化物;再其次,用去離子水超聲清洗,直至溶液呈中性;最后,用無水乙醇超聲清洗3次,將洗滌干凈的NF在60 ℃真空烘箱中干燥2 h,備用.
1.4.2 工作電極的制備
分別稱取5 mg催化劑和5 mg碳粉置于1.5 mL離心管中,再依次加入440 μL無水乙醇、60 μL Nafion溶液.將上述混合物超聲至糊狀.移取80 μL糊狀混合物均勻涂抹在導電載體NF上(涂覆面積約為1 cm × 0.8 cm),室溫下晾干,備用.
1.4.3 析氧反應性能測試
選用典型的三電極體系,將1.4.2所制備的電極作為工作電極,鉑片作為輔助電極,Ag/AgCl作為參比電極,測試溫度為25 ℃,電解液為1 mol/L的KOH.根據能斯特方程:ERHE=EAg/AgCl+0.059 2pH+0.197,將所有電位轉換為可逆氫電極(RHE).評估OER活性前,在0.1~0.6 V(vs.Ag/AgCl)電位窗口進行50圈循環伏安(CV)掃描,使催化劑活化.線性掃描伏安曲線(LSV)測試條件:掃描速率為5 mV/s;電勢窗口為0.9 ~1.6 V(vs.RHE).由LSV曲線計算可得塔菲爾(Tafel)斜率:η=a+blogJ(a為反應常數;b為Tafel斜率;J為電流密度).電化學阻抗譜(EIS)是在電壓1.46 V(vs.RHE)、頻率為100 kHz ~ 0.01 Hz、振幅為5 mV條件下獲得.通過雙層電容(Cdl)評估催化劑的電化學活性表面積(ECSA),在20、40、60、80、100 mV/s掃描速率下對非法拉第區域進行CV測試,利用0.125 V處的電流密度差(ΔJ/2)與掃描速率作圖,斜率即為Cdl值.采用計時電流法,在電流密度為0.01 A/cm2的條件下測定催化劑的長期穩定性.
2 結果與討論
2.1 基本表征
利用X射線衍射技術(XRD)表征In-MOF、In1Fe1-aMOF、In1Fe3-aMOF、In1Fe4-aMOF和Fe-aMOF的晶體結構,結果見圖1.

圖1 擬合的CPM-5材料、In-MOF、In1Fe1-aMOF、In1Fe3-aMOF、In1Fe4-aMOF和Fe-aMOF的XRD圖譜
由圖1可以看出:In-MOF衍射峰位置與單晶模擬的CPM-5[19]衍射峰位置相匹配,表明他們具有相同的晶體結構.In1Fe1-aMOF、In1Fe3-aMOF、In1Fe4-aMOF和Fe-aMOF的譜圖中僅出現幾個寬峰,并隨著In、Fe初始物質的量的比減小,峰強度也逐漸降低,說明樣品的結晶度較差.這可能是由于Fe3+的引入改變了局域電子分布,導致原始晶態In-MOF失去長程有序結構.
為了進一步探究催化劑的結構,采用紅外光譜儀對H3BTC、In-MOF、In1Fe1-aMOF、In1Fe3-aMOF、In1Fe4-aMOF和Fe-aMOF的官能團進行測定,結果如圖2所示.在H3BTC的紅外光譜圖中,1 723 cm-1和1 408 cm-1處的吸附峰歸屬于羧基(—COOH)伸縮振動.在In-MOF、In1Fe1-aMOF、In1Fe3-aMOF、In1Fe4-aMOF和Fe-aMOF的紅外光譜圖中,1 622 cm-1和1 372 cm-1處的吸附峰歸屬于去質子化羧基(—COO-)不對稱和對稱伸縮振動,同時在473 cm-1處檢測到新的振動吸收峰,表明金屬離子(In3+和Fe3+)與有機配體之間成功配位[20].通過ICP-AES測試定量分析催化劑中In3+和Fe3+的含量(見表1),隨著Fe(NO3)3·9H2O物質的量的增加,骨架中n(In)/n(Fe)比值逐漸降低.此外,In1Fe1-aMOF、In1Fe3-aMOF和In1Fe4-aMOF中n(In)∶n(Fe)分別為1∶1.2、1∶3.8、1∶5.1,略大于初始投料比,表明Fe3+與H3BTC間的配位能力更強,部分In3+并未參與配位而留在母液中.

圖2 H3BTC、In-MOF、In1Fe1-aMOF、In1Fe3-aMOF、In1Fe4-aMOF和Fe-aMOF的紅外光譜
通過掃描電子顯微鏡(SEM)和透射電子顯微鏡(TEM)對Fe-aMOF的微觀結構進行表征,結果如圖3所示.由圖3(a)可以看出Fe-aMOF呈球形,顆粒大小均勻,尺寸約為800 nm.由圖3(c)—圖3(e)可以看出:樣品中含有Fe、C和O三種元素,在整個材料中均勻分布,進一步證明Fe3+與有機配體之間成功配位,且催化活性位點分散良好.通過EDS分析可知Fe的質量分數約為50%.
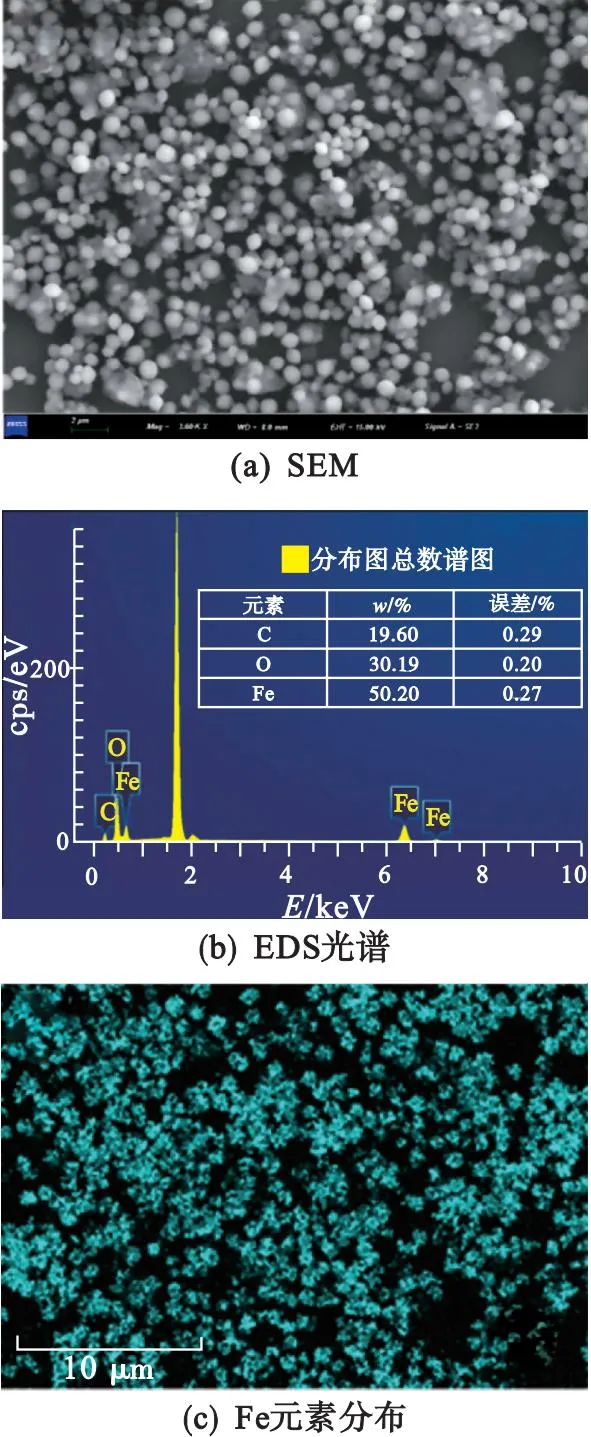
圖3 Fe-aMOF的SEM圖、EDS光譜及其相應的元素分布、TEM圖和SAED圖
在高分辨TEM圖像[圖3(f)]和相應的選定區域SAED[圖3(g)]中并未觀察到明顯的晶格條紋和可見的衍射環,進一步證明Fe-aMOF具有非晶態特征,與XRD結果相一致.由于非晶材料富含大量的缺陷位點,促使Fe-aMOF擁有更高的本征活性,有利于提高其電催化性能.
采用N2物理吸附探究In-MOF和Fe-aMOF的多孔結構,結果如圖4所示.由圖4可以看出:In-MOF和Fe-aMOF在低壓區快速吸附N2,說明兩種材料都存在微孔;與In-MOF相比,Fe-aMOF在高壓區平緩吸附N2,說明Fe-aMOF結構中還有部分介孔.圖4(a)顯示In-MOF的孔徑分布比較窄,主要集中于0.54~0.93 nm.而Fe-aMOF具有較寬的孔徑分布區間,除微孔外(0.52~2.00 nm),在2.00~10.00 nm間還出現了大量介孔[見圖4(b)],這進一步證明了Fe3+導致局部配位缺失,形成微-介孔結構.In-MOF和Fe-aMOF的孔結構信息見表2.Fe-aMOF和In-MOF的比表面積分別為506 m2/g和670 m2/g,Fe-aMOF的介孔孔容(0.190 cm3/g)遠高于In-MOF的介孔孔容(0.072 cm3/g),表明Fe-aMOF骨架中存在較多的介孔.這種微-介孔結構有利于提高活性比表面積和促進物質傳輸,從而提高Fe-aMOF的電催化性能.

表2 In-MOF和Fe-aMOF在77 K時的N2吸附結果
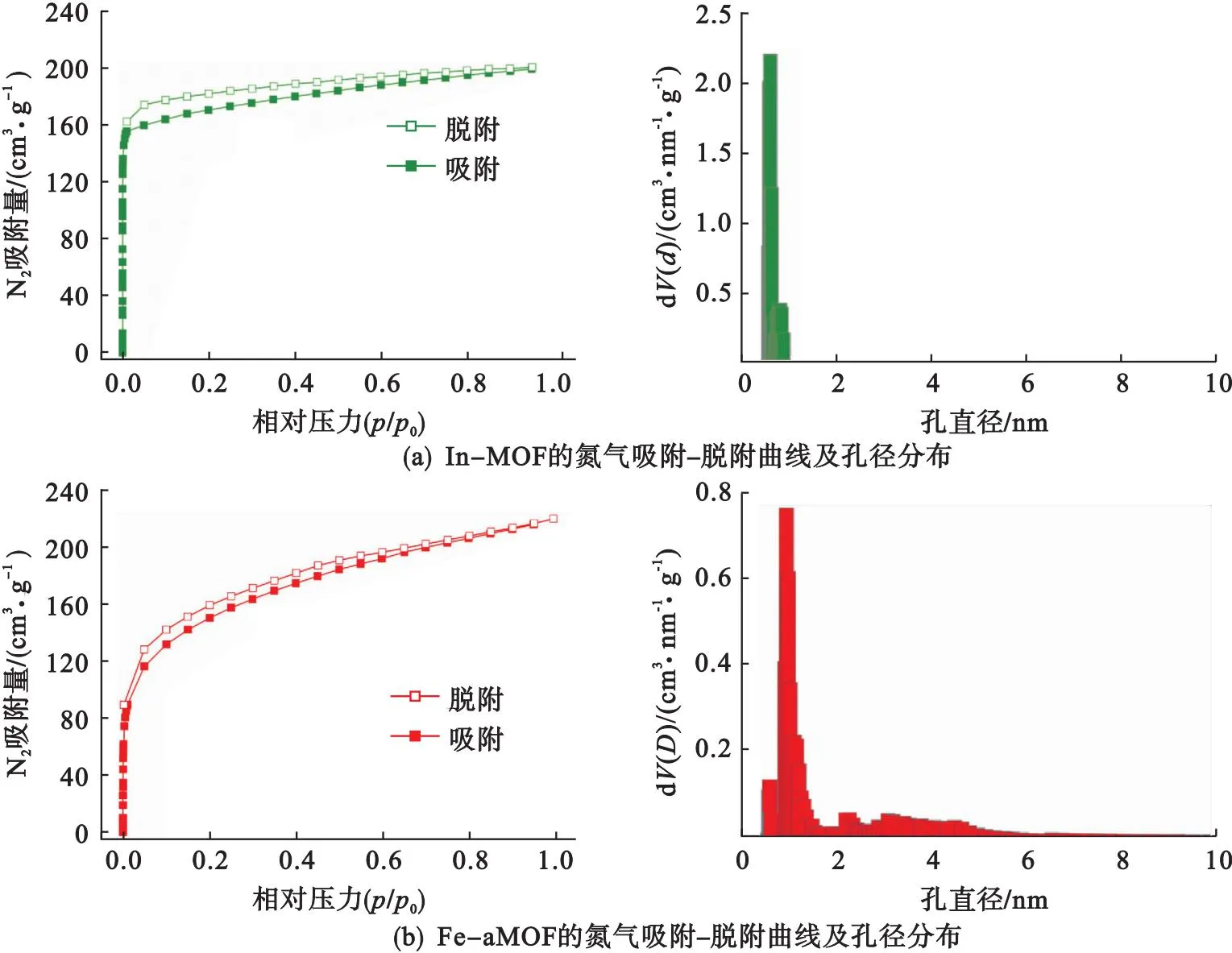
圖4 In-MOF和Fe-aMOF的氮氣吸附-脫附曲線及其相應的孔徑分布
2.2 電化學性能測試
將In-MOF、In1Fe1-aMOF、In1Fe3-aMOF、In1Fe4-aMOF和Fe-aMOF制成工作電極,采用典型的三電極體系,在1.0 mol/L的KOH溶液中探究其OER性能,結果如圖5所示.由圖5(a)可以看出:In-MOF在0.1 A/cm2時的過電位為341.9 mV;通過外來金屬離子誘導策略,隨著Fe3+含量的增加,OER性能也逐漸增強.Fe-aMOF表現出最高的OER活性,起始電位和過電位都很低:在電流密度為0.01 A/cm2時的過電位僅為258.2 mV,明顯低于In1Fe1-aMOF(301.1 mV)、In1Fe3-aMOF(279.6 mV)、In1Fe4-aMOF(269.8 mV)和空白NF(373.9 mV)的過電位;Fe-aMOF的過電位還低于大多數Fe基MOF催化劑[21-31]的過電位(見表3).為了深入研究電極材料的OER反應動力學,通過上述極化曲線,利用Tafel方程計算得出Tafel斜率.如圖5(b)所示,Fe-aMOF的Tafel斜率為45.4 mV/dec,低于In-MOF(68.8 mV/dec)、In1Fe1-aMOF(48.3 mV/dec)、In1Fe3-aMOF(46.9 mV/dec)、In1Fe4-aMOF(46.0 mV/dec)和空白NF(105.4 mV/dec)的Tafel斜率,表明Fe-aMOF具有最快的OER反應動力學.
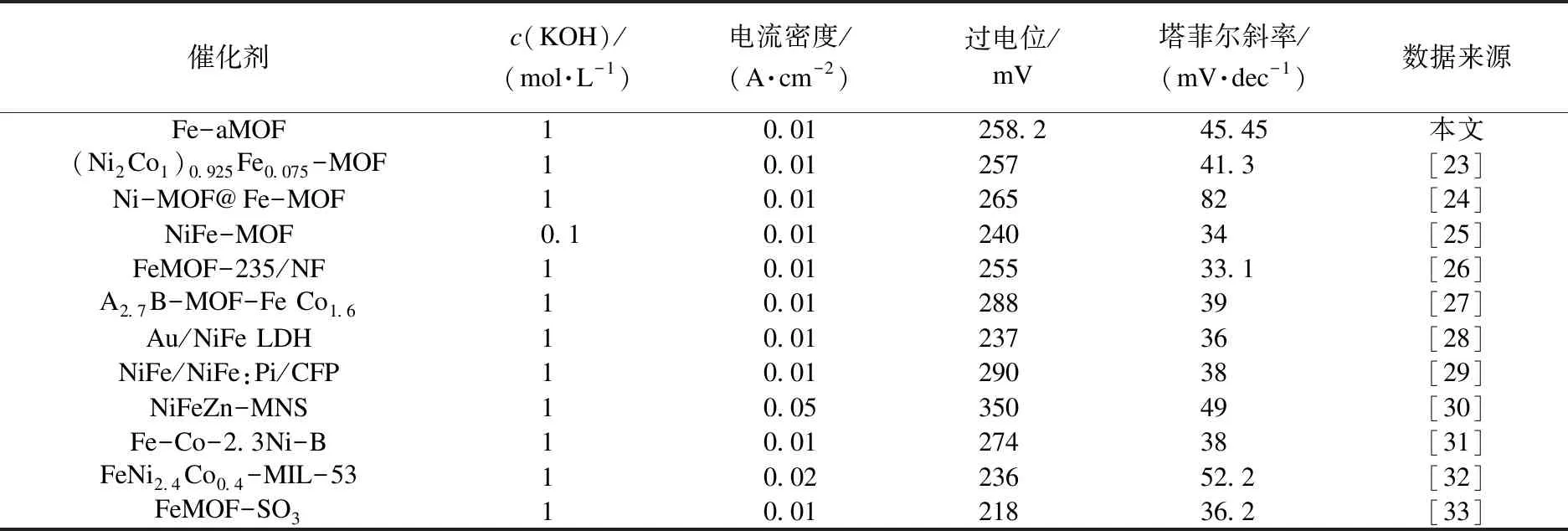
表3 Fe基電催化劑析氧性能比較

圖5 In-MOF、In1Fe1-aMOF、In1Fe3-aMOF、In1Fe4-aMOF、Fe-aMOF和NF的LSV曲線及其相應的Tafel圖
為了進一步探究Fe-aMOF OER性能提高的可能因素,評估了催化劑的電化學活性表面積(ECSA).由于電極的ECSA與其雙層電容(Cdl)成正比,因此可通過計算Cdl值的大小來反映催化劑的ECSA.圖6(a)—圖6(e)是In-MOF、In1Fe1-aMOF、In1Fe3-aMOF、In1Fe4-aMOF和Fe-aMOF在非法拉第區[電壓范圍0.1~0.15 V(vs.RHE)]、不同掃速下(20、40、60、80、100 mV/s)的CV曲線,在0.125 V(vs.RHE)處的ΔJ/2與掃描速率作圖,得到直線的斜率即為Cdl[見圖6(f)].Fe-aMOF的Cdl值為55.2 F/m2,高于In-MOF(29.5 F/m2)、In1Fe1-aMOF(34.1 F/m2)、In1Fe3-aMOF(47.2 F/m2)和In1Fe4-aMOF(50.7 F/m2)的Cdl值,表明Fe-aMOF的多孔性以及無定形結構賦予催化劑高的比表面積和更多裸露的活性位點,為電催化劑與電解質間提供有效的接觸面積.
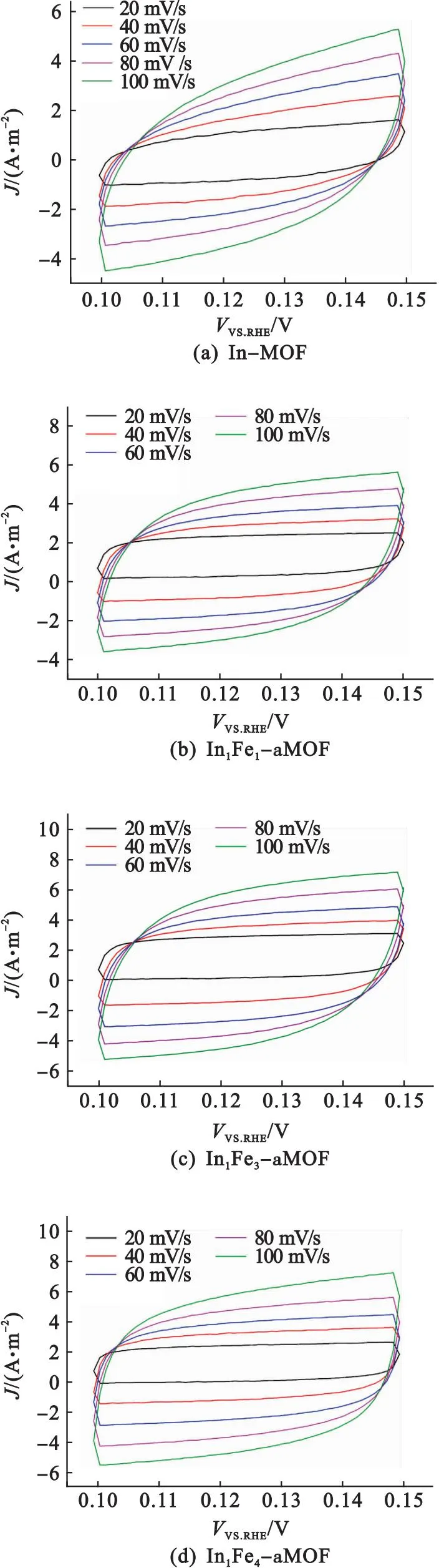
圖6 In-MOF、In1Fe1-aMOF、In1Fe3-aMOF、In1Fe4-aMOF和Fe-aMOF在不同掃描速率時的CV曲線及在非法拉第區0.125 V( vs.RHE)處ΔJ/2相對于掃描速率的函數
利用電化學阻抗(EIS)探索電催化劑在OER過程中的電荷傳輸能力.圖7為In-MOF、In1Fe1-aMOF、In1Fe3-aMOF、In1Fe4-aMOF和Fe-aMOF的Nyquist曲線,半圓的直徑可以粗略反映電催化劑阻抗的大小,即阻抗由小到大的電催化劑依次為Fe-aMOF、In1Fe4-aMOF、In1Fe3-aMOF、In1Fe1-aMOF和In-MOF.通過等效電路模型擬合可得到界面轉移電阻(Rct).其中:Fe-aMOF的Rct為1.04 Ω,遠小于In-MOF的10.19 Ω、In1Fe1-aMOF的1.93 Ω、In1Fe3-aMOF的1.66 Ω和 In1Fe4-aMOF的1.58 Ω,說明Fe-aMOF與電解質界面處的電荷轉移最快.此外,這些Rct值與電催化劑在電流密度為0.01 A/cm2時的過電位和Tafel斜率變化趨勢相同,可見Rct是影響OER活性的主要因素.
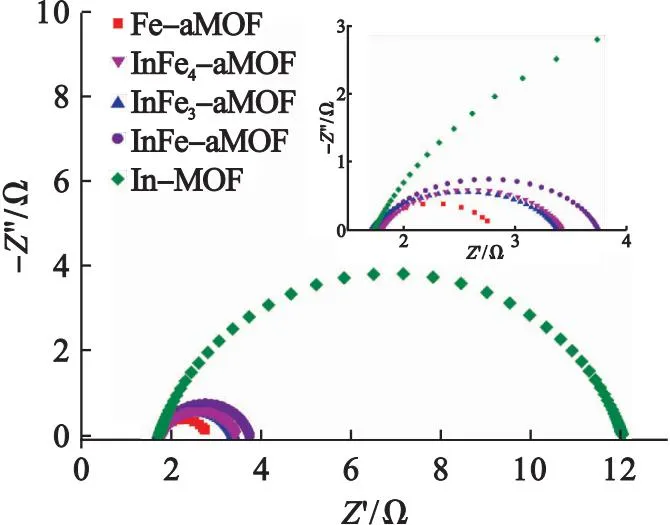
圖7 In-MOF、In1Fe1-aMOF、In1Fe3-aMOF、In1Fe4-aMOF和Fe-aMOF的Nyquist圖
除具有優異的OER活性外,電催化劑的穩定性也是評價能源轉換系統的一個重要標準.如圖8(a)所示,Fe-aMOF連續1 000圈循環伏安掃描后的LSV曲線與最初的LSV曲線非常相似,電流密度沒有明顯衰減.通過計時電流法評估Fe-aMOF的穩定性,結果如圖8(b)所示.

圖8 Fe-aMOF在進行1 000圈CV掃描前、后的LSV曲線和Fe-aMOF在0.01 A/cm2時的計時電流曲線
由圖8(b)可以看出:連續測試40 h內,Fe-aMOF的電流密度幾乎未出現較大波動,基本保持在0.01 A/cm2;連續測試超過40 h后,電流密度略微降低,可能是由電極表面產生大量氣泡使催化劑從載體上脫落導致的.利用ICP分別對連續測試40 h和110 h后的電解液進行分析,未檢測到金屬離子(Fe3+和Ni2+),以上結果說明導電載體和Fe-aMOF催化劑在1 mol/L的KOH溶液中皆具有良好的化學穩定性.
3 結 論
通過簡單溶劑熱法合成了一系列無定形的InxFey-aMOF材料,并采用SEM、TEM、XRD、FT-IR和ICP等對樣品的形貌、結構以及組成進行表征.研究結果表明:Fe3+的引入干擾了In3+與1,3,5-均苯三甲酸間配位,導致金屬中心-有機配體配位缺失.這種外來金屬離子誘導策略促使材料富有大量缺陷,暴露出更多的金屬活性位點,從而提高OER電催化活性.電化學結果表明:在1 mol/L的KOH電解液中,Fe-aMOF表現出最佳的電催化活性:電流密度達到0.01 A/cm2時,過電位僅需要258.2 mV,塔菲爾斜率為45.4 mV/dec,優于目前報道的多數晶態MOF材料.該研究缺乏理論計算,后續研究應將實驗結果與理論計算相結合,揭示催化劑結構與性能之間的構效關系.
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
