鄰苯二甲酸二-2-乙基己酯通過ROS/PTEN/PI3K/AKT軸誘導HD11細胞凋亡和程序性壞死
2024-01-10 03:05:20李廣興陳凱婷武夢林黃小丹
畜牧獸醫學報 2023年12期
李廣興,陳 陽,陳凱婷,武夢林,張 迪,黃小丹*
(1.東北農業大學動物醫學院,哈爾濱 150030;2.黑龍江省實驗動物與比較醫學重點實驗室,哈爾濱 150030)
鄰苯二甲酸二-2-乙基己酯[di(2-ethylhexyl)phthalate, DEHP]作為一種合成聚合物的增塑劑,已被廣泛應用于紡織品、醫療器械、電子產品和個人護理用品等方面[1]。而增塑劑會在材料中遷移并隨著時間的推移從材料中滲出,最終進入環境[2]。隨著DEHP的生產和使用量越來越大,其釋放在環境中的含量也呈逐年上升趨勢[3]。由于DEHP不斷地釋放到環境中,從而通過飲食,呼吸和皮膚接觸進入人體,進而危害人體健康,這引起了人們對其安全性及其對人體健康的潛在影響的一些擔憂[4]。廣泛的研究表明,DEHP對人和動物都有有害影響,可誘導生殖毒性、神經毒性和心臟毒性,此外還有免疫毒性[5-8]。據報道,在哮喘小鼠模型中,DEHP增加了總IgE水平,此外,BALF中酸性粒細胞數量和肺中酸性粒細胞陽離子蛋白(ECP)明顯增加[9]。研究人員發現DEHP可以改變骨髓中B細胞的成熟和分化[10]。此外,DEHP使小鼠巨噬細胞發生極化,M1極化降低,而M2極化增強,提示DEHP可以影響巨噬細胞的體內免疫反應,顯著降低其腫瘤預防能力[11]。DEHP持續灌胃45 d,會導致鵪鶉脾受損,組織病理學觀察脾組織中可見脾小體邊界和細胞間隙增大[12]。大量研究表明,DEHP等環境污染物造成的細胞損傷與氧化應激有關[13-14]。值得注意的是,DEHP通過增加ROS的產生,從而誘導氧化應激,增加Bax/cytochrome-c/Caspase-3的表達水平,抑制BCL-2的表達,啟動大鼠顆粒細胞的線粒體凋亡途徑[15]。此外,氧化應激損傷通過激活細胞程序性壞死途徑,增加壞死相關基因TNF-α、RIPK1、RIPK3、MLKL和JNK的表達,介導雞腎細胞程序性壞死[16]。
PTEN/PI3K/AKT通路參與了細胞生長、凋亡以及與環境有關的多個細胞過程,并與氧化應激的產生密切相關[17]。多項研究表明,當細胞發生氧化應激時,會刺激ROS的過度產生,進而激活PTEN的表達,從而抑制PI3K/AKT表達,這是導致細胞損傷的主要途徑[18-19]。PTEN/PI3K/AKT通路是細胞存活的保護通路,而氧化應激也常常通過調節PTEN/PI3K/AKT通路而導致細胞損傷。抑制miR-21能上調卵巢癌細胞中PTEN表達,阻斷PI3K/AKT通路的活性,從而促進細胞凋亡[20]。此外,不同劑量的CPF暴露草魚肝細胞,PTEN通過其脂質磷酸酶活性作用于PI3K/AKT通路,阻斷該通路活性從而導致細胞凋亡和壞死[19]。腫瘤壞死因子(TNF)誘導的細胞程序性壞死是受到PI3K/AKT的抑制,結果發現,RIP1、RIP3和MLKL的表達水平升高,表明抑制PI3K/AKT通路可激活細胞壞死通路,最終導致細胞程序性壞死[21]。值得注意的是,雙酚A(BPA)暴露通過上調Bax、caspase-3、caspase-9、RIP1、RIP3和MLKL的表達,下調BCL-2表達,加重了缺硒雞腎細胞凋亡和程序性壞死,PI3K/AKT的負調控在此過程中發揮著重要作用[22]。此外,硒減輕了鎘暴露通過抑制PI3K/AKT途徑導致的凋亡和程序性壞死,改善了鯉魚脾淋巴細胞的損傷[23]。
綜上所述,氧化應激調控PTEN/PI3K/AKT通路,在細胞凋亡和壞死中發揮著重要作用。此外,DEHP是一種有害的環境污染物。有研究表明,DEHP可引起細胞氧化應激和降低細胞中PI3K/AKT的表達水平,最終誘導細胞損傷[24-25]。然而,目前尚未有關于DEHP暴露通過ROS/PTEN/PI3K/AKT引起HD11細胞凋亡和程序性壞死的研究。因此,本試驗通過建立DEHP體外暴露模型,通過吖啶橙/澳化乙錠(AO/EB)染色法和流式細胞術檢測凋亡及壞死信號,氧化應激試劑盒檢測氧化與抗氧化指標,qRT-PCR和Western blot檢測PTEN/PI3K/AKT信號通路,凋亡相關基因(Bax、Caspase-3、Caspase-9和BCL-2)以及壞死相關基因(RIPK1、RIPK3和MLKL),探討其在免疫毒性進展中的具體機制。本試驗結果將為進一步探索塑化劑的免疫毒性機制提供新思路。
1 材料與方法
1.1 主要試劑
DEHP(純度≥99.0%,MCE,美國)。CCK8試劑盒、BCA試劑盒(碧云天生物技術公司,上海,中國)。青霉素、鏈霉素、胰酶(Gibco,美國)。胎牛血清(BioInd,上海,中國)。辣根過氧化酶標記的羊抗兔/鼠二抗(Abmart,上海,中國)。
1.2 HD11細胞培養
HD11細胞在RPMI-1640培養基(含10%胎牛血清、100 IU·mL-1青霉素、100 μg·mL-1鏈霉素)中,于37 ℃、飽和濕度條件下,含5%CO2的培養箱中培養。根據細胞生長狀況,每1 d傳代1次。將HD11細胞接種到每孔細胞密度為2×106的96孔板中。每孔加入細胞懸液100 μL,待細胞融合度達70%~80%時進行DEHP處理。DEHP暴露濃度分別為0、30、60、90、120、150、180、210、240 μmol·L-1DEHP進行處理。DEHP暴露24 h后,每孔加入10 μL CCK8,隨后在溫箱中繼續孵育2 h,用酶標儀測定450 nm處的吸光度。按照說明書的公式計算不同濃度DEHP處理細胞后的細胞活力,并以折線圖的形式呈現不同濃度DEHP暴露HD11細胞后的細胞活力。使用軟件SPSS分析半數抑制濃度(IC50)和95%置信區間。
1.3 HD11細胞分組
根據DEHP暴露對HD11細胞24 h的IC50,應用細胞存活率為≥80%的30 μmol·L-1DEHP為最低劑量,最終在DEHP致HD11細胞凋亡和程序性壞死及氧化應激作用的研究中,本試驗用不同濃度的DEHP (0、30、60、90 μmol·L-1)處理HD11細胞24 h分別命名為對照組(C組)、低劑量組(L組)、中劑量組(M組)和高劑量組(H組)。
1.4 氧化應激相關指標的測定
按照南京建成試劑盒說明書對LMH細胞進行相應處理。用酶標儀在525 nm/532 nm/520 nm/550 nm/412 nm波長下測定ROS/MDA/T-SOD/T-AOC/GSH-PX的吸光度。
1.5 AO/EB染色和流式細胞術
1.5.1 AO/EB染色 每100 μL細胞懸液加入4 μL AO/EB混合液(AO∶EB=1∶1),在室溫避光培養5 min后,將其放入熒光顯微鏡觀察形態學改變,然后拍照記錄。根據以下的公式計算細胞凋亡指數。凋亡指數=凋亡細胞數/(正常活細胞數+凋亡細胞數+壞死細胞數)×100%。壞死指數=壞死細胞數/(正常活細胞數+凋亡細胞數+壞死細胞數)×100%。
1.5.2 流式細胞術 加入195 μL Annexin V-FITC結合液重懸細胞,再避光加入5 μL Annexin V-FITC 和10 μL碘化丙啶,混勻,避光于室溫下溫育20 min。轉移至新的流式玻璃管中,加入1×binding buffer 400 μL,輕彈混勻。用流式細胞術(NovoCyte,Aceabio,USA)和FlowJo7.6.1軟件檢測細胞凋亡率和壞死率。
1.6 總RNA分離及實時熒光定量PCR分析
使用總RNA試劑盒II(天根生物技術有限公司,北京,中國)中提取HD11細胞總RNA,反轉錄為cDNA,以β-actin為內參基因。qRT-PCR在LightCycler@480系統(羅氏,瑞士)上進行。引物序列分別見表1。熒光定量PCR反應條件:94 ℃預變性2 min;94 ℃變性15 s,60 ℃退火15 s,72 ℃延伸30 s,共45個循環;以GAPDH的Ct值為內參,采用相對定量分析2-ΔΔCt對得到的Ct值進行數據分析。
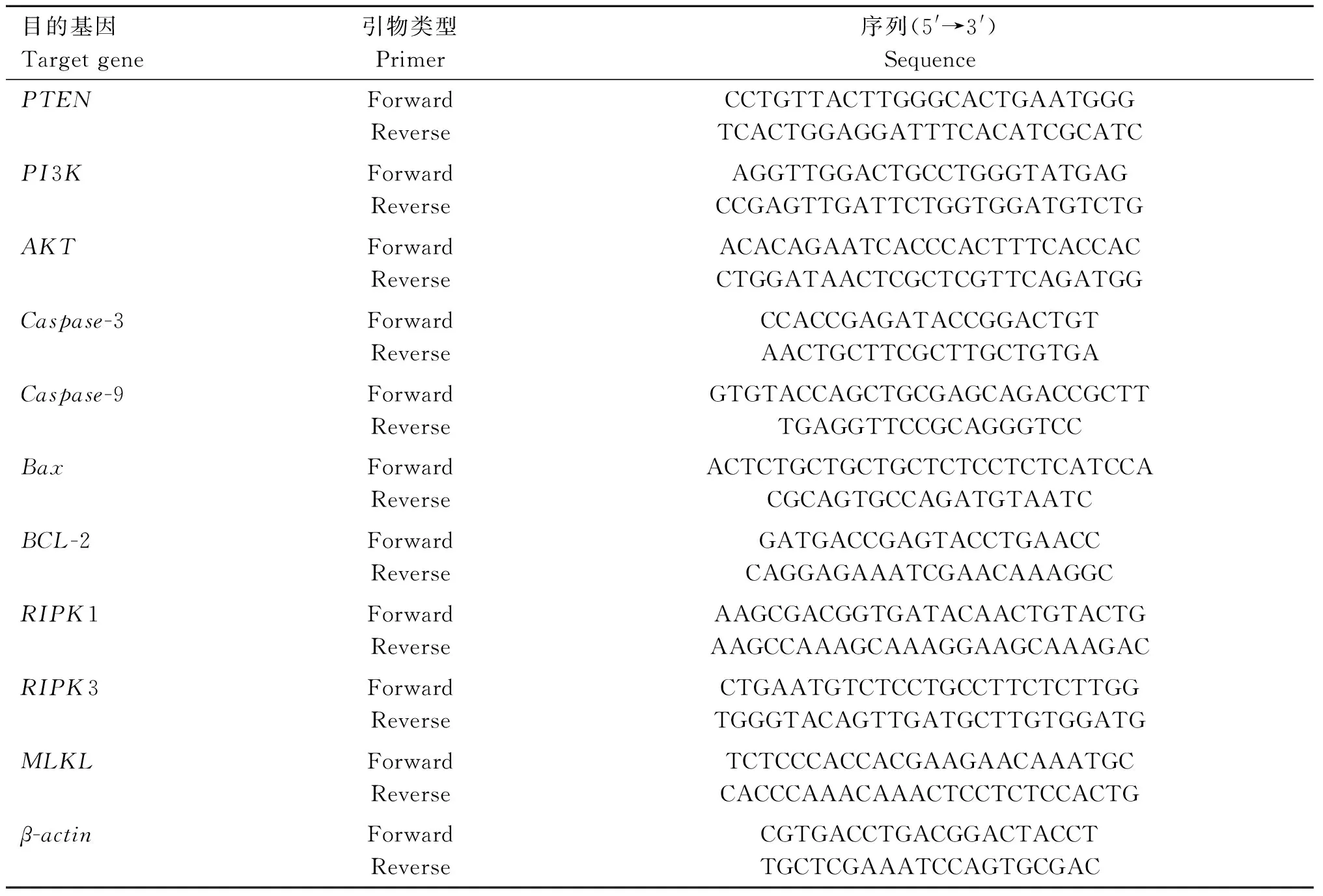
表1 雞 qRT-PCR 的基因序列Table 1 Gene-special primers used for qRT-PCR in chicken
1.7 Western blot檢測
采用BCA試劑盒檢測蛋白濃度。將電泳后分離的蛋白電轉至硝酸纖維素膜上,5%脫脂奶37 ℃封閉2 h,隨后,將纖維膜在4 ℃的一抗中孵育過夜。用TTBS洗滌3次,每次15 min,接著在室溫下進行二抗孵育時長為2 h,然后用TTBS洗滌3次,每次15 min。ECL發光壓片顯色,以GAPDH作為內參,掃描并分析圖像,并計算相對表達量。本研究使用的抗體如下:PTEN(1∶1 000,萬類,沈陽),PI3K(1∶1 000,萬類,沈陽),AKT(1∶1 000,萬類,沈陽),BCL-2(1∶1 000,萬類,沈陽),Bax(1∶1 000,萬類,沈陽),Caspase3(1∶1 000,萬類,沈陽),Caspase9(1∶1 000,萬類,沈陽),RIPK1 (1∶1 000,Abmart,上海),RIPK3(1∶1 000,Abmart,上海),MLKL(1∶1 000,Abmart,上海),GADPH(1∶1 000,Abmart,上海),HRP標記的山羊抗兔IgG(1∶5 000,ZSGB-B)。
1.8 統計分析
采用SPSS(17版; SPSS,Chicago,IL,USA)進行統計學顯著性分析。用GraphPad(版本7.0,GraphPad Software inc. ,San Diego,CA,USA)進行統計學分析,P<0.05具有統計學意義。在每種情況下至少進行3個獨立的試驗,并以平均值(SEM)的“平均值±標準誤差”表示數據。
2 結 果
2.1 DEHP暴露對HD11細胞活力的影響
為了明確DEHP對HD11的半數抑制濃度,用0、30、60、90、120、150、180、210、240 μmol·L-1DEHP處理HD11細胞24 h,隨后進行CCK-8檢測細胞活力,如圖1所示,結果表明,隨著DEHP濃度的升高,細胞活力呈濃度依賴式下降,DEHP顯著抑制了HD11細胞的細胞活力。DEHP對HD11細胞24 h的IC50為180.644 μmol·L-1(95%置信區間:160.725~208.598 μmol·L-1)。
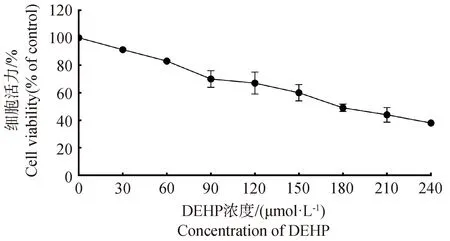
圖1 DEHP暴露對HD11細胞活力的影響Fig.1 Effect of DEHP exposure on HD11 cell viability
2.2 DEHP暴露對HD11細胞氧化應激指標的影響
為了評估DEHP暴露對HD11細胞氧化應激的影響,因此,本試驗檢測了相關的氧化應激指標。結果如圖2所示,與對照組相比,DEHP處理組的ROS與MDA表達水平顯著升高,并且與DEHP濃度成正比(P<0.01)。此外,與對照組相比,T-AOC表達水平顯著降低,抗氧化物酶T-SOD,GSH-PX活性顯著降低(P<0.01),結果表明,DEHP可誘導HD11細胞發生氧化應激,從而導致HD11細胞損傷。
2.3 AO/EB染色和流式細胞術分析
為了證實DEHP暴露可誘導HD11細胞發生細胞凋亡和程序性壞死,用AO/EB染色和流式細胞術檢測了不同暴露劑量下HD11細胞的細胞凋亡和程序性壞死比例。如圖3A所示,AO/EB染色后,熒光顯微鏡可區分出4種細胞形態:活細胞,核染色質著綠色并呈正常結構;早期凋亡細胞,核染色質著綠色呈固縮狀或圓珠狀;晚期凋亡細胞,核染色質為橘紅色并呈固縮狀或圓珠狀;非凋亡的死亡細胞,核染色質著橘紅色并呈正常結構。在熒光圖像中,隨著DEHP暴露劑量的升高,L組、M組和H組的凋亡細胞和壞死細胞比例逐漸增加。此外,Annexin VFITC-PI熒光標記法探究細胞凋亡和壞死的具體數量。如圖3C所示,流式細胞儀數據顯示,壞死細胞比例5.1%增加到16.2%(第一象限),第二、三象限顯示,凋亡細胞比例5.8%增加到19.8%,AO/EB和流式細胞術結果共同表明,DEHP以濃度依賴性的方式增加了凋亡細胞和壞死細胞的比例。
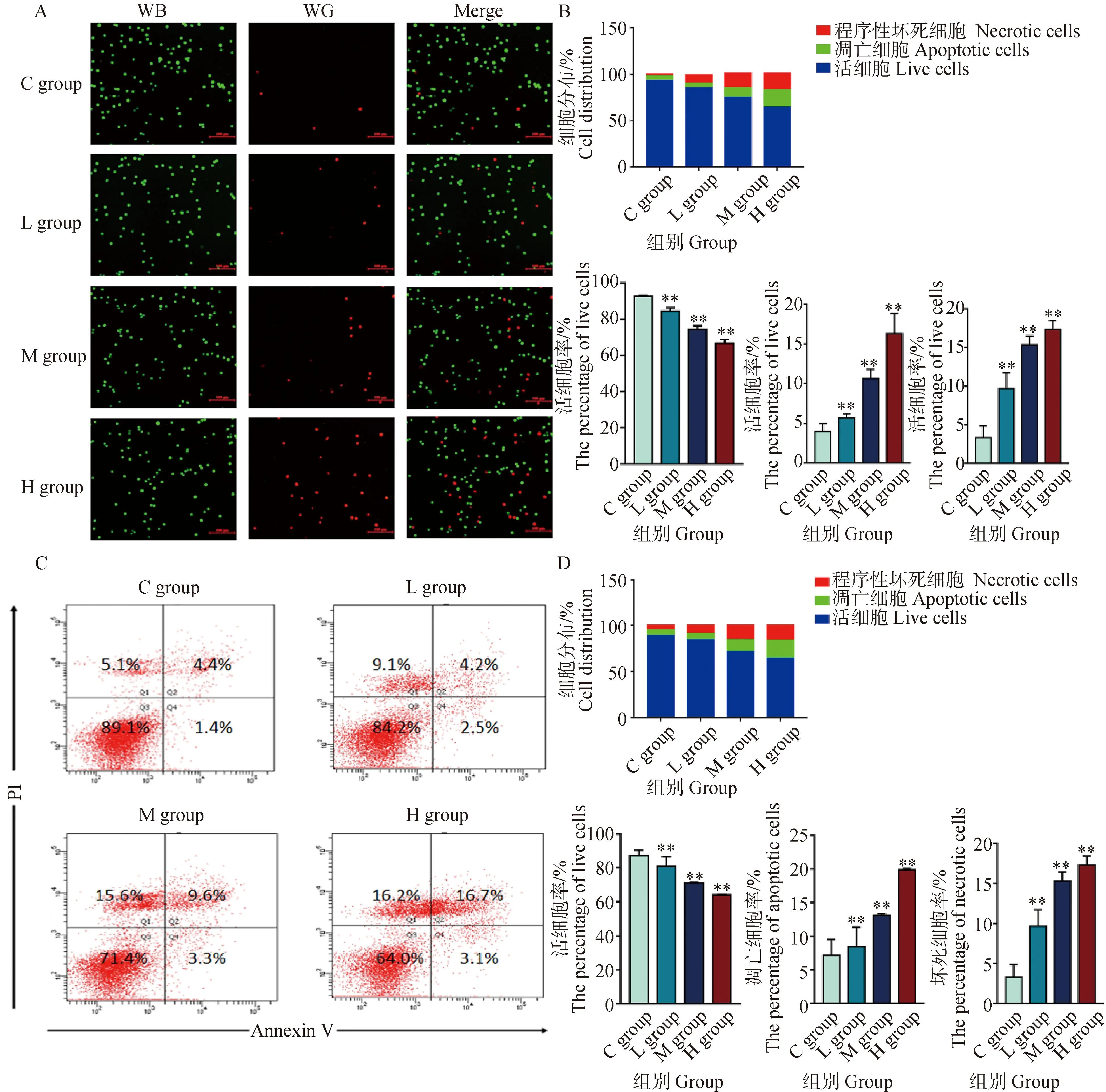
A. AO/EB染色(比例尺,100 μm);B. Image J 熒光定量強度分析;C.流式細胞術分析;D.流式細胞術定量分析。與對照組(0 μmol·L-1)相比,**. P<0.01A. The AO/EB fluorescence staining (scalebar, 100 μm); B. The AO/EB fluorescence intensity quantitative analysis by ImageJ; C. The results of flowcytometry analysis; D. The quantitative results of flowcytometry. Compared with C group (0 μmol·L-1),**. P<0.01圖3 HD11細胞凋亡檢測結果Fig.3 Cell apoptosis assay results for HD11 cells
2.4 DEHP暴露對HD11細胞PTEN/PI3K/AKT通路相關基因表達的影響
為了探索DEHP誘導的HD11損傷過程是否與PTEN/PI3K/AKT通路有關,本試驗檢測了DEHP暴露24 h后,HD11細胞中PTEN/PI3K/AKT信號通路相關基因的表達情況。如圖4所示,與對照組相比,DEHP暴露組PTEN的mRNA和蛋白水平顯著升高。DEHP暴露組PI3K和AKT mRNA及蛋白表達明顯降低(P<0.01)。結果表明,DEHP能增強HD11細胞中PTEN的表達,進而抑制PI3K和AKT的表達,且這種作用隨著DEHP劑量的增加而加強,提示DEHP能有效調控HD11細胞內PTEN/PI3K/AKT通路的活性。
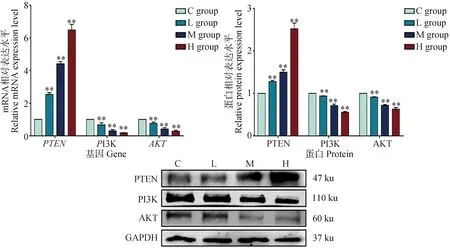
與對照組(0 μmol·L-1)相比,**. P<0.01Compared with C group (0 μmol·L-1),**. P<0.01圖4 PTEN/PI3K/AKT通路相關基因mRNA及蛋白表達變化Fig.4 Changes of mRNA and protein expression of PTEN/PI3K/AKT pathway related genes
2.5 DEHP暴露對HD11細胞線粒體凋亡相關基因表達的影響
為了研究DEHP對HD11細胞凋亡的影響,進一步檢測了凋亡通路相關因子Caspase 9、Caspase 3、BCL-2和Bax的mRNA和蛋白表達。如圖5所示,與對照組相比,DEHP暴露組抗凋亡基因BCL-2的mRNA和蛋白表達顯著降低,而Caspase 9、Caspase 3和Bax的mRNA和蛋白表達顯著增加(P<0.01)。結果表明DEHP暴露誘導HD11細胞發生線粒體凋亡。
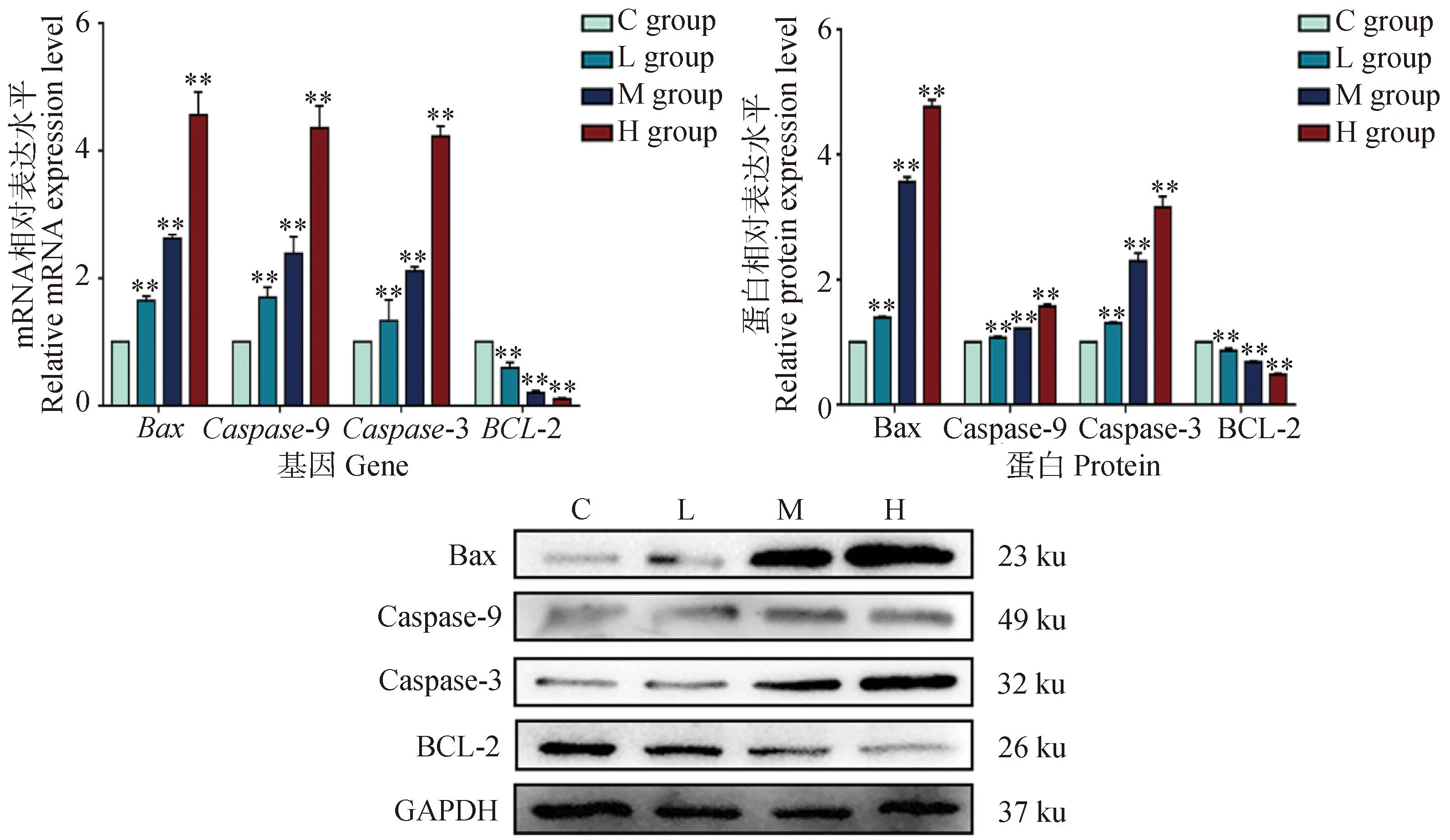
與對照組(0 μmol·L-1)相比,**. P<0.01Compared with C group (0 μmol·L-1),**. P<0.01圖5 細胞凋亡相關基因mRNA及蛋白表達變化Fig.5 Changes of mRNA and protein expression of apoptosis related genes
2.6 DEHP暴露對HD11細胞程序性壞死相關基因表達的影響
為了驗證DEHP暴露24 h后細胞內的壞死通路被激活,分析了程序性壞死信號通路相關基因RIPK1、RIPK3和MLKL的mRNA和蛋白表達,由圖6所知,當DEHP暴露時,與對照組相比,RIPK1、RIPK3和MLKL的mRNA和蛋白表達水平均顯著升高(P<0.01)。表明DEHP暴露誘導HD11細胞發生程序性細胞壞死。
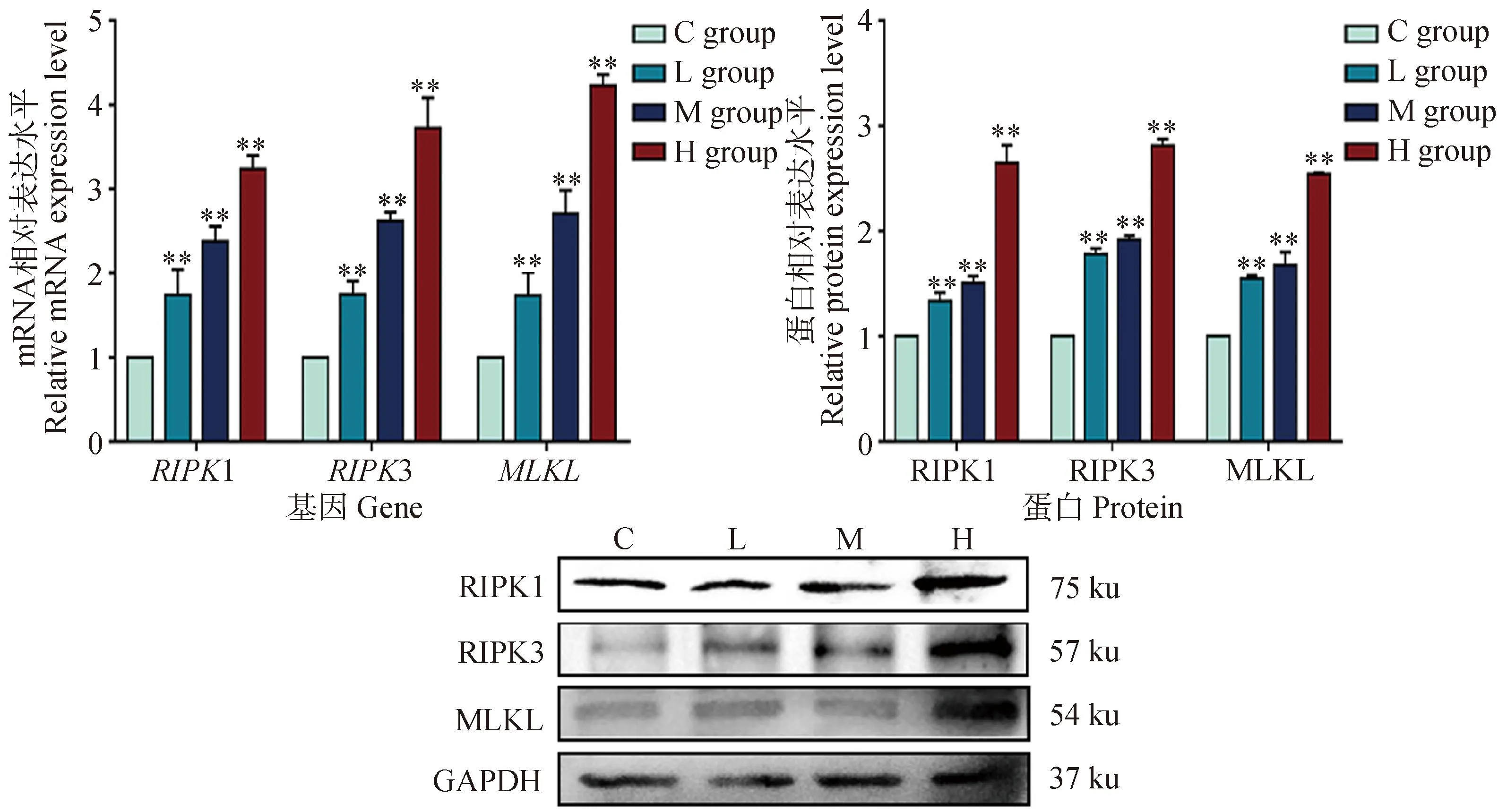
與對照組(0 μmol·L-1)相比,**. P<0.01Compared with C group (0 μmol·L-1),**. P<0.01圖6 程序性壞死相關基因mRNA及蛋白表達Fig.6 Changes of mRNA and protein expression of necroptosis related genes
2.7 主成分分析和相關性分析
為了進一步探究DEHP暴露引起的HD11細胞凋亡和程序性壞死與PTEN/PI3K/AKT通路是否有關,利用PCA對上述指標進行了分析,由圖7A所知,所有檢測基因均位于三維結構中,PC1、PC2、PC3分別代表不同的3個組成部分,占比為94.6%、3.7%和0.8%。通過Pearson檢驗各基因之間的相關性。從結果中發現PTEN、PI3K、AKT通路以及凋亡和程序性壞死相關基因主要集中在PC1,且PI3K、AKT、BCL-2之間呈正相關,與其他因子呈負相關,相關分析結果如圖7B所示,顯示相關分析的結果與PCA基本一致。提示PTEN負調控PI3K/AKT通路進而促進DEHP誘導的HD11細胞凋亡和程序性壞死。
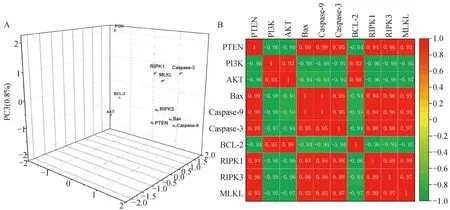
圖7 主成分分析(A)及相關性分析(B)Fig.7 Principal component analysis (A) and correlation analysis (B)
3 討 論
DEHP作為無處不在的存在于食品和自然環境中的污染物,可誘發免疫系統損傷和氧化應激損傷[26-27]。研究人員發現暴露于不同劑量的DEHP,會產生大量ROS,最終導致大鼠卵巢顆粒細胞發生線粒體凋亡[15]。此外,PTEN/PI3K/AKT通路與毒性作用機制密切相關,參與了細胞凋亡和程序性壞死通路的激活[19]。本研究探討了DEHP是否誘導雞巨噬細胞凋亡和程序性壞死及其可能機制,本試驗研究了DEHP調控ROS/PTEN/PI3K/AKT通路在DEHP誘導雞巨噬細胞凋亡和程序性壞死中的作用,結果表明,DEHP暴露引起了HD11細胞程序性壞死和細胞凋亡,發生了氧化損傷。此外,PTEN/PI3K/AKT通路被激活,并存在劑量效應關系。誘導雞巨噬細胞凋亡和程序性壞死。本研究為比較醫學研究提供理論基礎。
生物體內活性氧(ROS)生成量是一個動態變化過程,與許多內外因素有關,它直接決定是否發生氧化應激,而這也決定了細胞存活和死亡的不同調節[28]。在正常生理條件下,ROS主要被細胞內的抗氧化系統清除,從而使其維持在較低的水平參與細胞信號調節。然而,當細胞內抗氧化系統被抑制或者抗氧化系統與ROS的產生之間失去平衡時,氧化應激就會產生,當ROS過度產生便會導致氧化損傷[29]。研究顯示,環境中的許多化學物質均可誘導細胞及器官產生氧化應激[30-32]。不同濃度的H2O2作用于HepG2細胞時,ROS水平顯著增加并與H2O2呈劑量效應關系[33]。此外,維生素E和姜黃素單獨作用均能有效減弱鄰苯二甲酸二丁酯(DBP)引起的脾氧化應激損傷[34]。DEHP被證明可以誘導多種細胞和器官產生氧化應激,如斑馬魚肝細胞、人子宮內膜基質細胞以及大口黑鱸幼魚的肝和脾[35-38]。與以往研究相似,本研究結果顯示,隨著DEHP暴露水平的增加,ROS和MDA水平顯著升高,T-SOD、GSH-PX活性顯著降低。此外,暴露于DEHP的HD11細胞中T-AOC水平顯著降低。結果提示,DEHP暴露后,HD11細胞內ROS的過量生成和細胞抗氧化能力的降低,導致抗氧化酶系統的破壞,導致細胞發生氧化應激。
DEHP暴露可誘導細胞毒性損傷,這與PTEN/PI3K/AKT信號通路的激活密切相關。例如,研究發現DEHP通過上調PTEN的表達,進而下調AKT的表達,從而抑制人胚胎干細胞增殖,促進細胞周期停滯,并誘導細胞凋亡[39]。DEHP暴露通過抑制PI3K/AKT信號通路,誘導心肌細胞毒性[40]。還有研究發現DEHP可通過激活氧化應激和下調PI3K/AKT信號通路誘導骨骼肌細胞凋亡和程序性壞死[41]。與以往研究相似,本試驗結果顯示,DEHP暴露后,HD11細胞發生氧化損傷,DEHP處理組PTEN基因的mRNA和蛋白表達相應升高,PI3K和AKT表達降低。值得注意的是,BCL-2和RIPK1是PI3K/AKT信號轉導通路的重要下游靶點,通過抑制PI3K/AKT表達,進而使BCL-2低表達和RIPK1高表達來實現凋亡和壞死[42-43]。研究人員發現,用50和500 mg·(kg·d)-1DEHP處理小鼠35 d,會使BCL-2蛋白低表達,使Bax蛋白高表達,進而Bax與BCL-2形成異二聚體從而啟動信號通路,導致線粒體損傷,最終誘導小鼠睪丸啟動內質網應激誘導生殖細胞凋亡[44]。此外,500 μmol·L-1DEHP可以上調RIPK1、RIPK3和MLKL表達水平,最終導致心肌細胞發生程序性壞死[45]。與以往研究相似,本研究發現,不同濃度DEHP暴露于HD11細胞導致BCL-2的表達水平顯著降低,Bax、Caspase-3、Caspase-9表達顯著增強,啟動了細胞線粒體凋亡途徑;同樣,RIPK1、RIPK3和MLKL的表達顯著升高,說明細胞程序性壞死通路也被激活(圖8)。
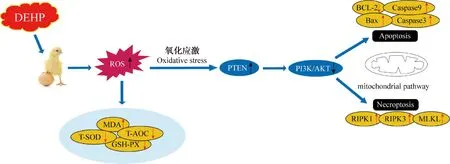
圖8 圖形摘要Fig.8 Graphical abstract
4 結 論
本研究首次提供了DEHP暴露引起HD11細胞凋亡和程序性壞死證據。發現DEHP可抑制HD11細胞活力,導致氧化應激反應增強,進而激活了PTEN/PI3K/AKT信號通路,促進了HD11細胞凋亡和程序性壞死,并且HD11細胞的損傷程度與DEHP濃度成劑量依賴性。這些發現為闡明DEHP對禽類中毒的分子機制提供了研究基礎,為尋找切實有效地治療DEHP所致的HD11細胞損傷藥物提供了思路。本研究對DEHP的毒理學研究、生態環境保護及動物和人類健康具有重要意義。
猜你喜歡
體育科技文獻通報(2022年3期)2022-05-23 13:46:54
天津外國語大學學報(2021年3期)2021-08-13 08:32:18
遼金歷史與考古(2021年0期)2021-07-29 01:06:54
世界科學技術-中醫藥現代化(2020年2期)2020-07-25 02:05:56
科技傳播(2019年22期)2020-01-14 03:06:54
民用飛機設計與研究(2019年4期)2019-05-21 07:21:24
汽車工程學報(2017年2期)2017-07-05 08:13:02
西南軍醫(2016年6期)2016-01-23 02:21:19
新疆醫科大學學報(2015年10期)2015-12-26 12:33:30
癌變·畸變·突變(2015年3期)2015-02-27 06:15:09
