Li/SF6 燃燒反應路徑及機理研究
2024-01-13 12:17:20溫伯堯王起源孫成珍駱政園白博峰
水下無人系統學報 2023年6期
關鍵詞:模型
溫伯堯 ,王起源 ,孫成珍 ,宗 瀟 ,駱政園 ,白博峰 *
(1.西安交通大學 動力工程多相流國家重點實驗室,陜西 西安,710049;2.中國船舶集團有限公司 第705 研究所,陜西 西安,710077)
0 引言
由堿或堿土金屬(如Li、Al、Mg 等)和強氧化劑組成的金屬燃料系統相比于碳氫燃料系統,具有能量密度高、體積小、低噪聲等優點,在能源動力領域具有非常顯著的優越性[1-3]。其中,由于Li與SF6是比較理想的金屬燃料和氧化劑,具有反應產熱高、反應過程穩定、反應物易存放、無氣態產物排放、安全以及無噪聲等優點[4-6],因此Li/SF6通常被用作熱源,可與不同熱機搭配構成閉式循環熱動力系統。Li/SF6燃燒反應放熱劇烈,產物十分復雜,反應機理涉及化學反應、相變、高溫氣體裂解、多相流動和湍流等復雜物理化學過程。揭示Li/SF6燃燒微觀反應機理對于構建金屬燃料反應動力學模型、實現燃燒過程高效組織具有十分重要的指導和實際意義。
Li/SF6作為最具代表性的金屬燃料系統之一,國內外學者對其進行了大量研究,主要包括反應機理研究、實驗觀測及數值仿真等。20 世紀60 年代,荷蘭研究人員發表了多組分在不互溶相中的熱力學模型和熱化學數據、Li/SF6浸沒燃燒的多流體湍動模型[7];1992 年美國愛荷華大學的Lyu等[8-9]提出了一個基于守恒標量方法和界面分析的Li/SF6燃燒預測模型。我國相關研究起步較晚,朱強[10-11]計算了Li/SF6反應產物Li2S 的比熱、焓和熵等熱力學數據,并將結果擬合成多項式,這些數據在Li/SF6閉式循環動力系統研究中得到了應用;張文群等[12-14]通過建立Li/SF6氣液浸沒燃燒反應相平衡計算的非線性約束優化方法,計算了反應的平衡產物、溫度和密度等,給出了Li 初始溫度的影響,并利用Gibbs 自由能最小法研究了Li/SF6氣液浸沒燃燒反應中混合分數與溫度、密度和平衡產物質量分數等的關系。目前,對金屬Li 的相關反應機理鮮有研究,化學反應動力學數據更是缺乏。
Li/SF6燃燒反應實驗主要用于觀測射流尺寸、溫度變化以及產物分布。Avery 和Facth[15]通過實驗獲得了反應區尺寸、擴展速率及射流中心線處的溫度分布,發現高金屬密度的射流導致蒸汽相的出現,進而發生凝結和擴散;Parnell 等[16]利用X 光照相技術觀測了反應區尺寸、穩定性及擴展速率,結果表明射流長度與熔融鋰濃度有關;Hsu 等[17]實驗觀測了點火及燃燒過程,將其分為點火、火焰傳播、穩定燃燒和熄火4 個階段;鄭邯勇等[18]利用X 光高速動態成像技術觀測了Li/SF6浸沒噴射反應過程,測量了SF6在液態鋰中的浸沒噴射長度和寬度,建立了描述SF6在液態鋰中浸沒噴射長度的半經驗關系式。李維維等[19]開展了Li/SF6的小功率鍋爐反應器快速啟動試驗,研究了點火后氧化劑和冷卻水進入時序、流量匹配和啟動劑產物等對化學反應和換熱的影響。由于燃燒實驗成本大、測量困難,而單獨金屬顆粒的燃燒無法體現反應組分間相互作用,Li/SF6浸沒燃燒實驗難度極大。
數值仿真可以預估實驗難測量參數,節省研究費用,縮短研究周期,指導反應器結構設計及參數優化,有利于提高系統可靠性和經濟性,因此廣泛應用于Li/SF6燃燒反應研究。Chan 等[20]應用均質流(homogeneous flow,LHF)模型仿真發現反應距離很短,但射流深度大,同時研究了Li 局部低溫對流動結構的影響;Chan 等[21]研究發現,與LHF 模型相比,多流體模型仿真得到的射流深度與實驗數據更為接近;Dahikar 等[22]研究了噴嘴直徑及布置方式、射流氣體速度、熔融Li 溫度對噴射尺寸的影響,發現噴嘴水平放置對反應更有利,可以得到穩定的射流長度;Chen 等[23]建立了燈芯式燃燒的數學模型,闡明雷諾數是影響強制對流燃燒的重要參數,氣流速度增加使得總燃燒速率增加;Gulawani 等[24]研究發現傳質系數與湍動能及湍流擴散率有關,基于多流體模型的浸沒噴射流型、射流尺寸及溫度等的結果與實驗數據更為相符。
盡管數值仿真在重現實驗過程和數據方面取得了進展,現有針對低功率密度Wick 燃燒和高功率密度浸沒燃燒仿真的燃燒反應模型誤差仍較大,這一方面是因為未考慮輻射換熱,導致溫度場的準確性降低,另一方面是缺乏化學反應機理認識,Li/SF6反應活化能、反應熱、反應速率以及擴散系數等熱/動力學參數的設置缺乏理論指導。目前對于Li/SF6反應的中間步驟認識不清,只能采用基元反應代替反應過程,計算誤差較大。因此通過分子仿真的手段來揭示金屬燃料反應機理是非常有必要的。基于反應力場(reactive force field,ReaxFF)的分子動力學仿真完全由體系勢能推動,計算過程中無須預設反應路徑,非常適用于分析反應體系的微觀機理。
ReaxFF[25-26]能夠在原子水平上描述反應物和產物在不同條件下的結構變化和化學反應等過程,通過分析反應過程中組分的變化可以得到反應路徑及中間產物等重要信息。為了揭示Li/SF6燃燒的分布反應機理,文中結合ReaxFF 反應分子動力學仿真和第一性原理計算方法研究了熱源啟動過程中Li 和SF6的微觀反應過程,分析了主要反應物和產物組分的動態演化特性,獲得其主要反應路徑及反應熱,為構建或驗證Li/SF6燃燒反應動力學模型提供機理認識和理論基礎。
1 分子動力學仿真模型及方法
1.1 仿真模型
首先,采用Materials Studio 軟件構建Li 和SF6單分子的幾何模型,并進行結構優化;其次,采用Amorphous Cell 模塊構建包含多個Li 和SF6分子的周期性盒子(初始尺寸為7×7×7 nm3)作為仿真模型,一定數目的Li 和SF6分子在盒子內隨機分布;再次,根據初始溫度條件合理設定反應物的初始密度以避免原子重疊,通過改變盒子中Li/SF6分子數目及比例來控制反應物組分的質量比;最后,利用Forcite 模塊在等溫等壓系綜(NPT)和正則系綜(NVT)下對特定溫度(500 K)和壓力(0.1 MPa)條件下的反應體系進行50 ps 的非反應動力學平衡過程仿真,從而完成分子動力學仿真模型(見圖1,圖中紅色為S 原子,藍色為F 原子;綠色為Li 原子)的構建。

圖1 Li/SF6 反應的分子動力學仿真模型Fig.1 Molecular dynamics simulation model of Li/SF6 reaction
1.2 仿真方法
上述分子動力學仿真模型隨后采用LAMMPS軟件中的ReaxFF 程序包開展分子動力學仿真,所用力場參數為Islam 等[27]擬合開發的C/O/H/F/Li/S體系力場參數。該計算力場中C/H/O/S 和Li 參數,以及F 參數是基于已有文獻中的力場參數,并經量子化學計算進行了額外的力場訓練,能夠準確描述鍵解離、Li-F 相互作用,以及其他分子與Li 和F 的結合。計算過程中采用周期性邊界條件和微正則系綜(NVE),時間步長為0.25 fs,采用Berendsen 方式控制體系溫度,溫度阻尼系數設置為10 fs。仿真工況如表1 所示。每個工況進行3 次平行仿真以獲得更好的統計學結果。仿真完成后,采用鍵級截斷的方法判斷原子成鍵及反應產物,統計分析原子軌跡、關鍵產物及化學鍵等相關信息,追蹤特征產物組分隨反應過程的動態演化規律,進而分析總反應中各個分反應的內在聯系及其主次規律。
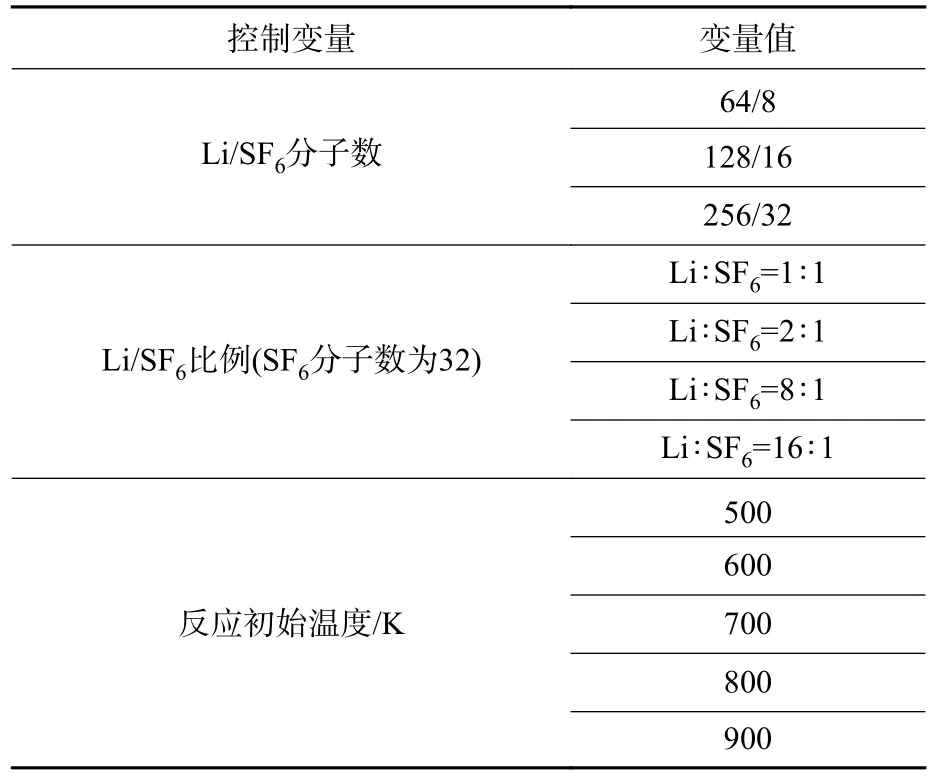
表1 Li/SF6 反應仿真工況Table 1 Simulation conditions of Li/SF6 reaction
1.3 第一性原理計算
從量子力學理論出發的第一性原理計算方法,根據原子核和電子相互作用的原理及其基本運動規律,近似處理后直接求解薛定諤方程,能夠得到物質的基態性質。因此文中采用第一性原理計算Li/SF6反應過程中各物質的基態性質。
采用Material Studio 軟件中的DMol3 模塊[28]對反應過程的熱力學性質進行分析計算。通過得到體系的總電子和離子能量Etotal以及焓值的修正值Htotal之后,計算可得反應物/產物總的焓值H=Etotal+Htotal。體系的總電子和離子能量Etotal包含于體系內能U中,內能由電子、振動、平移和旋轉分量組成,即
在反應焓值計算過程中,首先建立單獨的分子模型并采用DMol3 模塊對分子結構進行幾何優化,得到分子的基態結構;之后使用統計力學獲得簡正模的振動頻率,并對分子進行振動分析計算,即可獲得式(1)中的各項數值;利用DMol3 中的analysis 功能計算得到重要的熱力學性質,從中讀取修正焓值Htotal與溫度之間的函數關系,得到在特定溫度T下的焓值修正值Htotal;計算得到各個分子的焓值之后,根據反應熱的計算公式求得總的反應熱。
反應熱?H可以利用反應物焓值Hreactatns與生成物焓值Hproducts之差進行計算,即
2 仿真結果與分析
2.1 主要反應路徑分析
為了解Li/SF6反應的主要路徑,文中設置了多組不同SF6和Li 分子數目的工況進行對比分析,在當前仿真盒子尺寸條件下,SF6和Li 分子數分別為8 和64 時便能獲得相對穩定的結果。圖2(a)展示了特定工況(反應物分子數SF6和Li 比例為8∶64,初始溫度為500 K)下仿真系統的最終構象,可以看出反應生成了固體產物LiF 和Li2S。圖2(b)給出了該工況下主要反應物和產物組分隨仿真時間的變化情況。從反應過程中各個組分的分子數變化及反應分子動態視圖可知,反應的起始過程為SF6分子S-F 鍵的斷裂,LiF 是反應初始時刻的主要產物;隨著反應的進行,多余的Li 反應形成Li2或與S 成鍵形成Li2S;反應后期,LiF 分子間發生反應結合生成Li2F2或Li3F3。由此可以給出Li/SF6反應的主要反應路徑(見圖2(b)): SF6→S+6F、Li+F→LiF、2Li→Li2、Li2+S→Li2S、2LiF→Li2F2。
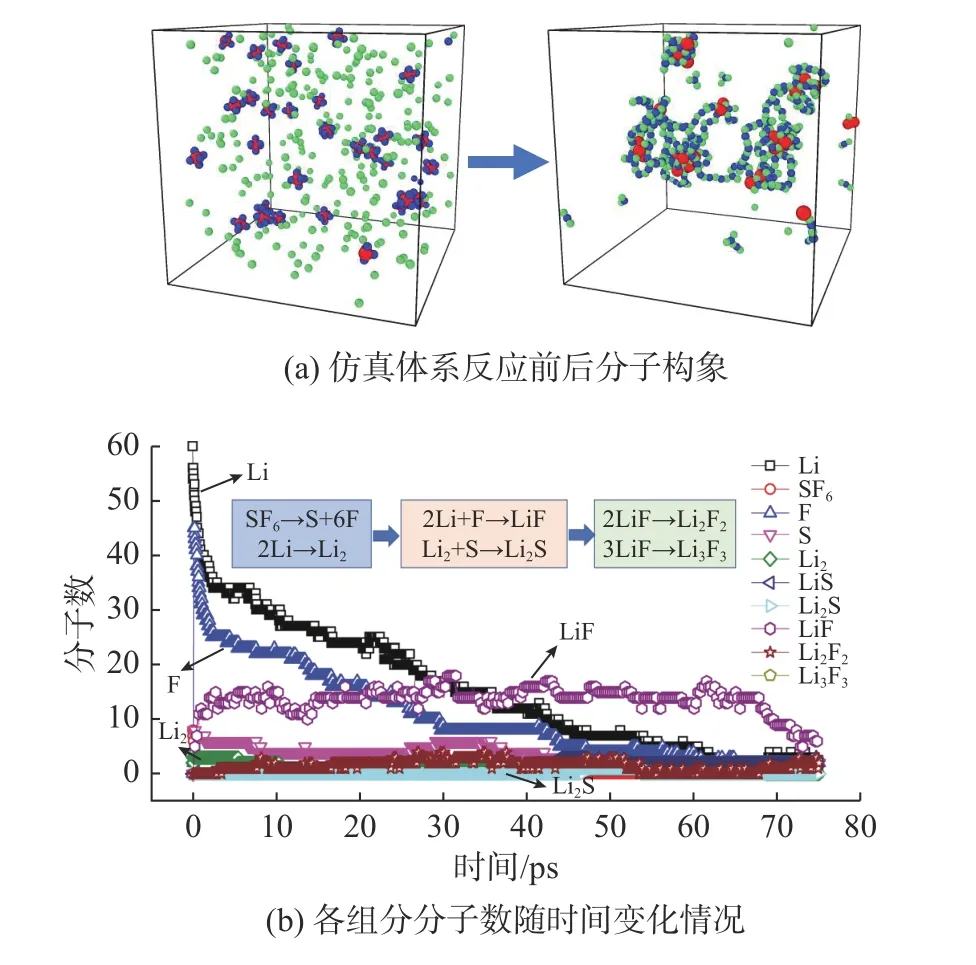
圖2 Li/SF6 反應前后系統構象及體系中組分分子數隨時間變化情況Fig.2 Conformation of Li/SF6 system before and after reaction and variation of molecule number with time
由于溫度是影響反應的關鍵因素,因此文中首先研究了不同初始溫度對反應過程的影響。圖3展示了反應物分子數SF6和Li 比例為8∶64 時,不同初始溫度下Li 和LiF 分子數隨時間的變化情況。由圖可知,初始溫度對反應初始階段影響較小,這是因為反應初始階段主要為SF6分解,SF6分解速率在當前仿真溫度(500~900 K)相差不大。但是隨著初始溫度增加,Li 消耗速率增加,同時反應接近平衡時的Li 剩余比例也較低,表明更多的Li 與其他原子發生反應,總反應也進行得更為徹底。這一點從圖中LiF 的分子數變化也可以推測得到。隨著反應初始溫度增加,更多的Li 與F 反應生成LiF,因此初始溫度越高,反應趨近平衡時的LiF 分子數越多。由于文中重點關注反應的初始階段,溫度對反應的影響放在后續研究中,這里不再贅述。

圖3 不同初始溫度下Li 和LiF 分子數隨仿真時間變化情況Fig.3 Variation of Li and LiF molecule number with simulation time under different initial temperatures
文中同時研究了反應物濃度對Li/SF6反應過程的影響。圖4 展示了在相同仿真盒子尺寸、反應物比值(SF6∶Li=1∶8)和初始溫度(500 K)條件下Li、F、LiF 這3 種主要組分隨時間的變化情況。由圖可知,隨著反應物濃度增加,反應進行得越快,反應接近或達到平衡所需的時間越短,這是由于隨著反應物濃度增加,分子間距離縮短,反應物分子間的碰撞概率增加,使得反應加快。產物LiF 的變化情況則較為復雜,不僅受到前序反應(SF6分解)的影響,還與LiF 的后續反應有關: 反應初期,LiF 分子數隨反應物濃度的增加而增大;隨著反應的不斷進行,LiF 分子數越大,碰撞概率越大,不斷結合反應生成Li2F2,因此反應物濃度越高的體系中LiF 分子數反而越小。

圖4 不同反應物濃度下Li、F 和LiF 分子數比例隨時間變化情況Fig.4 Variation of proportion of Li,F,and LiF molecule number with time under different reactant concentrations
文中也分析了不同仿真盒子尺寸和溫度(500 K)條件下,維持SF6分子數不變,通過改變反應物Li 的分子數來分析反應物比例對反應過程的影響。圖5 展示了反應物Li 和主要產物LiF 數目隨反應時間的變化情況。由圖5 可知,隨著Li 分子數增加,其與S、F 原子發生碰撞概率增加,反應物Li 消耗速率增加;反應產物LiF 在反應初始階段隨Li 分子數增加而增加,反應后期多余的Li 與LiF 繼續反應生成Li2F2和Li3F3,反而導致LiF 分子數減少,這也從側面證明了上述主要反應路徑的合理性。

圖5 不同反應物比例下Li 和LiF 分子數占比隨時間變化情況Fig.5 Variation of proportion of Li and LiF molecule number with time under different reactant proportions
2.2 反應速率計算
通過分析反應物Li 的濃度變化來獲得總反應的反應速率。在容積不變的反應容器里,反應速率v(mol·L-1·s-1)定義為單位體積內反應進度(ε=Δn/ν)對時間的變化率,即
式中:V為體積;v 為化學計量數;n為物質的量;c為物質的量濃度。
圖6 給出了不同反應條件下Li/SF6總反應速率隨時間的變化情況。由圖可知,反應速率與反應物濃度或分子數呈正相關關系,在相同反應容積下,反應物濃度越大,反應物分子間距離越小,碰撞概率越高,因此反應速率越大;Li 在很短時間內(≈10 ps)被反應消耗掉,反應速率隨著Li 的消耗迅速減小后趨于穩定;隨著反應物比例(或Li 分子數)增大,反應速率幾乎呈同比例線性增加,因為隨著Li 分子數增加,Li 與其他原子碰撞發生反應的概率也增加;反應初始溫度對總反應速率的影響較小,但從LiF 分子數變化可以推斷初始溫度對分步反應的反應速率是有影響的。
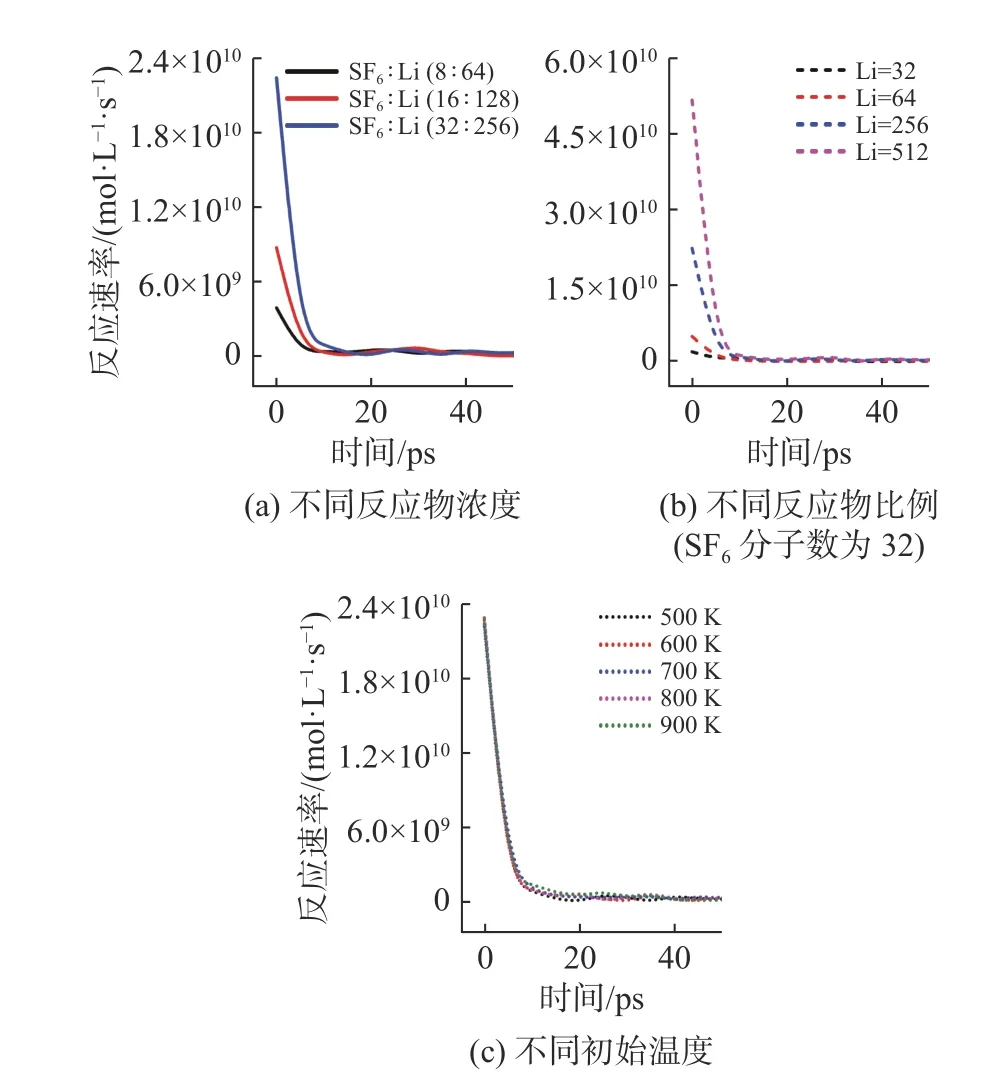
圖6 不同反應條件下反應速率隨時間變化情況Fig.6 Variation of reaction rate with time under different reaction conditions
2.3 反應熱計算
根據上述確定的分反應,采用第一性原理計算獲得每個分反應對應的放熱量。反應過程中各分子的能量折線圖和分子示意圖如圖7 所示。

圖7 反應過程中反應物的能量折線圖及分子示意圖Fig.7 Energy line diagram and molecular diagram of reactants in reaction process
上述分析得到的反應SF6→S+6F、Li+F→LiF、2Li→Li2、Li2+S→Li2S 為總反應的分反應,而2LiF→Li2F2為總反應產物的雙分子結合反應,根據蓋斯定律[29]可得總反應的反應熱計算公式為
反應2LiF→Li2F2的反應熱計算公式為
因此整個化學反應過程的反應熱為上述2 部分反應熱的總和。需要注意的是生成1 個Li2F2分子需要2 個LiF 分子,而總反應中共生成6 個LiF 分子,因此需要調整反應熱的系數,即
由上式可知,為了計算反應的生成熱,需要計算得到各個反應物/產物的焓值。采用第一性原理計算獲得了各個反應物/產物分子的焓值,如表2所示。
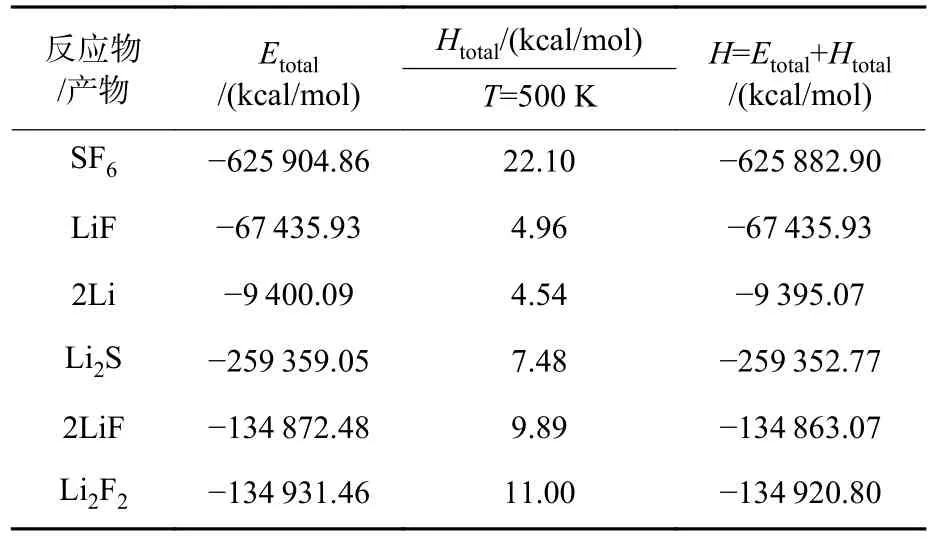
表2 Li/SF6 反應過程中反應物/產物焓值計算結果Table 2 Calculation results of reactant/product enthalpy value during Li/SF6 reaction
根據上表計算所得焓值,可通過以下公式計算得到Li/SF6反應的反應熱:
根據反應物/產物標準焓值計算得到的反應熱為-2 318 kJ/mol,文獻中實驗測得的反應熱為-2 621 kJ/mol[18],均與文中計算結果接近。計算得到的反應熱與理論值及實驗值之間的誤差主要來源于仿真溫度、壓力等條件與實際反應條件的差異,以及仿真采用的反應力場與實際反應過程的吻合程度。表明通過分子仿真獲得主要反應路徑,然后再基于第一性原理計算得到復雜反應過程的反應熱具備一定的可靠性。
3 結論
文章采用ReaxFF 反應分子動力學仿真方法研究了Li 和SF6微觀反應過程,獲得產物分布和動態演化特性,以及其主要反應路徑,然后結合第一性原理計算得到了主要反應路徑的反應熱,主要結論有以下幾個方面。
1) Li/SF6反應的起始過程為SF6分子S-F 鍵的斷裂,LiF 是反應初始時刻的主要產物;隨著反應的進行,多余的Li 形成Li2與S 成鍵形成Li2S;反應后期,LiF 分子間結合反應形成Li2F2。由此判斷Li/SF6反應主要路徑為: SF6→ S+6F、Li+F →LiF、2Li→Li2、Li2+S→Li2S、2LiF→Li2F2。
2) 獲得了反應物濃度、比例以及初始溫度對反應速率的影響規律: 在相同容積條件下,反應速率大小與反應物濃度呈正相關,濃度越大,反應物分子間距離越小,發生碰撞概率越高,因此反應速率越大;隨著反應物比例增大,反應速率幾乎呈同比例線性增加,因為Li 與其他原子碰撞概率增加;初始溫度對總反應速率的影響較小,但對分步反應的反應速率是有影響的。
3) 采用第一性原理計算獲得Li/SF6反應主要路徑或分步反應對應的放熱量,進而獲得Li/SF6反應的反應熱為-2 216.7 kJ/mol,表明通過分子仿真獲得主要反應路徑,然后結合第一性原理計算得到反應熱是可行的,可用于復雜燃燒反應機理揭示及反應熱計算。
總之,文中建立了能夠描述Li 和SF6微觀反應過程的分子仿真方法,結合第一性原理計算實現了Li 和SF6反應的主要路徑判斷及反應熱計算,分析了反應物濃度、反應物比例及初始溫度對總反應速率的影響規律,揭示了Li 和SF6燃燒反應的微觀機制。研究結果為揭示復雜燃燒反應的微觀機理和反應熱計算提供了有效手段,獲得了Li/SF6燃燒反應的反應熱、反應速率等關鍵動力學及熱力學參數,為構建或驗證Li/SF6燃燒反應動力學模型提供了機理認識。
但是文中一方面尚未揭示反應溫度對反應效率以及反應路徑的影響規律和機制,后續研究需著重針對此方面展開。另一方面,由于Li/SF6燃燒反應過程非常復雜,受溫度差異、時空尺度差異以及力場極化等因素的影響,當前基于ReaxFF 力場的分子動力學仿真模型還難以直接服務于燃燒反應微觀模型的建立,筆者正通過結合傘狀采樣方法開展加速分子動力學仿真,嘗試建立介尺度燃燒反應分子模型;同時,鑒于微觀反應與宏觀多相流動在時間和空間尺度上的巨大差異,當前微觀尺度的仿真研究還難以考慮宏觀尺度流體動力學的影響,亟需發展兼顧微觀反應和宏觀流動的介觀尺度或多尺度仿真技術,結合統計物理方法,建立一個可被移植到宏觀仿真的微觀模型與工具,在微觀反應機理與宏觀燃燒過程間搭建起溝通的橋梁,從而更好地指導燃燒反應器的優化設計與應用。
猜你喜歡
童話王國·奇妙邏輯推理(2024年5期)2024-06-19 16:03:38
網絡安全與數據管理(2022年1期)2022-08-29 03:15:20
導航定位學報(2022年4期)2022-08-15 08:27:00
中學生數理化·中考版(2022年8期)2022-06-14 06:55:24
新世紀智能(數學備考)(2021年9期)2021-11-24 01:14:36
成都醫學院學報(2021年2期)2021-07-19 08:35:14
新世紀智能(數學備考)(2020年9期)2021-01-04 00:25:14
中學生數理化·七年級數學人教版(2020年10期)2020-11-26 08:24:50
數學物理學報(2020年2期)2020-06-02 11:29:24
光學精密工程(2016年6期)2016-11-07 09:07:19
