烯丙基活化制備大孔強陽離子交換介質及其在蛋白分離中的應用
2024-01-13 00:00:00薛彥曉侯恒揚公丕勝靳海波趙嵐何廣湘張榮月
分析化學 2024年12期

關鍵詞強陽離子交換層析介質;烯丙基活化;配基密度;吸附性能;分離純化
隨著生物制藥技術的發展,生物制品的制備規模越來越大,對高通量分離純化的需求越來越多。層析技術由于其易放大、操作溫和而逐漸成為主流技術之一。其中,離子交換色譜(IEC)具有良好的生物相容性和溫和的洗脫模式,并且能夠在分離過程中最大限度地保持生物分子的活性,因而在蛋白質類藥物的分離純化過程中得到了廣泛應用[1-3]。離子交換介質的性能常決定其分離純化工藝的優劣,目前以多糖為基質的層析介質應用較多,然而這類介質本身質地軟的性質(耐壓小于0.3MPa)限制了其在高通量分離過程中的應用。聚合物基質具有機械強度高、表面化學性質易控制等特點,因此以該類基質制備的層析介質備受關注[4-5]。
烯丙基活化是制備陽離子交換介質的常用方法。該方法采用烯丙基溴化物或烯丙基縮水甘油醚對基質進行活化,溴化物或縮水甘油酯基在堿性環境下與基質發生反應,而相對惰性的烯丙基一端不參與反應,然后利用烯丙基的不飽和化學鍵進一步衍生其它功能單體。該方法通常在水溶液中進行,其活化試劑(烯丙基溴化物或烯丙基縮水甘油醚)廉價易得,鍵合配體不易脫落,因而被廣泛應用。Burton等[6]采用烯丙基活化的方法制備了一種混合模式介質,該介質對乳糜蛋白的吸附可取代高配基密度的無水碳基二咪唑介質。他們在另一項工作中[7]采用自由基加成的方式制備基質,并以烯丙基對其活化,制備了陽離子交換介質和親和介質。烯丙基活化法多用于瓊脂糖基質的層析介質制備中,而用于聚合物基質層析介質的制備方法的研究較少。
本研究采用大孔聚甲基丙烯酸酯類微球為基質制備了一種強陽離子交換介質,其基質微球富含環氧基團且易親水改性和功能衍生,同時其大孔結構有利于實現高通量分離[8]。聚丙烯酸酯類微球作為聚合物層析介質基質的優勢在于其表面親水性較好,同時相比于瓊脂糖凝膠基質具有較高的機械械強度(可耐受壓力達到1MPa以上)[9]。采用烯丙基活化方法,以烯丙基縮水甘油醚為活化試劑,偶聯小分子配基偏重亞硫酸鈉,制備得到大孔強陽離子交換介質,對烯丙基活化條件進行了優化,對配基密度的影響因素進行了系統考察,對不同配基密度的介質進行了性能評價,并將其用于模型分子溶菌酶的分離純化。本研究為大孔強陽離子交換介質的設計和應用提供了理論借鑒。
1實驗部分
1.1儀器與試劑
L5紫外可見分光光度計(上海儀電公司);SHA-BA水浴恒溫振蕩器(金壇榮華公司);EM-30plus臺式掃描電子顯微鏡(韓國COXEM公司);PPS-100蛋白純化系統(蘇州中科森輝公司)。
聚甲基丙烯酸縮水甘油酯與乙二醇二甲基丙烯酸酯共聚大孔微球(PGMA-EDMA,北京博爾賽譜生物科技有限公司);Na2S2O5和NaOH(分析純,國藥集團化學試劑有限公司);烯丙基縮水甘油醚(AGE,分析純)和溶菌酶(Lys,來源于雞蛋清)(北京華威銳科化工有限公司)。其它試劑均為國產分析純試劑;實驗用水為去離子水。
1.2大孔強陽離子交換介質的制備
制備路線如圖1所示,共分三步。第一步反應是在酸性條件下,將PGMA-EDMA微球的環氧基水解為鄰羥基。第二步進行烯丙基活化[10],具體過程如下:(1)將50g抽干的PGMA-EDMA微球加入到150mL0.5mol/L的H2SO4溶液中,置于50℃搖床上反應15h,制得水解完全的微球,以去離子水反復沖洗至中性,烘干備用;(2)將3g水解微球加入到裝有5mLNaOH(50%,m/V)的具塞錐形瓶內,再加入9mLAGE,搖勻,抽真空10min,在70℃水浴搖床上隔絕空氣反應18h,反應結束后,先用去離子水洗滌多次,然后用乙醇清洗至無乳白色液體,抽干即得烯丙基活化的微球。第三步反應是氧化所得烯丙基為磺酸基團,將3g抽干的烯丙基活化的微球和10mL1.5mol/L的Na2S2O5溶液加入到錐形瓶中,置于35℃空氣搖床上反應24h后,用去離子水清洗,再用砂芯漏斗抽干,即得大孔強陽離子交換介質。
1.3烯丙基密度測定實驗
烯丙基密度的測定參考文獻[11]的方法,計算公式如下:
其中,Q為烯丙基密度(mmol/g);C0為Na2S2O3標準溶液的濃度(mol/L);V0為滴定空白組所消耗的Na2S2O3標準溶液的量(mL);V為滴定微球所消耗的Na2S2O3標準溶液的量(mL);m為所取烯丙基活化微球的質量(g)。
1.4配基密度測定實驗
配基密度的測定根據文獻[12]的方法,采用酸堿滴定法測定,計算公式如下:
其中,N為離子交換容量(mol/L);N1為NaOH溶液濃度(mol/L);N2為HCl溶液濃度(mol/L);V1為所用NaOH溶液體積(mL);V2為滴定時所需HCl溶液體積(mL);V3為滴定微球體積(mL)。
1.5靜態載量(SBC)測定
緩沖液為20mmol/L磷酸鹽緩沖液(PB,pH=7.0;如無特殊說明,以下測試所用緩沖液皆為此溶液),模型蛋白為溶菌酶,吸附時間為6h,使用紫外可見分光光度計測量溶液在280nm波長處的紫外吸收,通過外標法計算出其對應的溶菌酶濃度C1。
其中,QSBC為單位體積介質靜態結合蛋白載量(g/L);C1為吸附后的蛋白濃度(g/L);C2為吸附前的蛋白濃度(g/L);V3為加入蛋白體積(mL);V4為所用介質體積(mL)。
1.6動態載量及回收率測定
動態載量及回收率均采用PPS蛋白純化系統進行測定。1mL層析柱(?7mm×2.5cm)用自制介質裝填,緩沖液為20mmol/LPB(pH=7.0),蛋白溶液為1g/L溶菌酶,洗脫液為含0.5mol/LNaCl的PB溶液,再生液為0.5mol/LNaOH,動態載量以10%流穿作為標準。回收率計算公式如下:
其中,R為蛋白回收率(%);A0為空載蛋白峰面積(即未連接色譜柱測定蛋白峰面積,作為100%回收標準);A1為洗脫蛋白峰面積。
1.7雞蛋清中溶菌酶的分離
采用自制介質對雞蛋清中的溶菌酶進行分離并用凝膠電泳對其純度進行測試,緩沖液為pH=8.0的20mmol/L的PB,洗脫液為含1mol/LNaCl的PB,采用5mL層析柱,以5倍柱體積上樣,上樣和洗脫流速均為1mL/min,采用等度洗脫。
2結果與討論
2.1微球形貌的表征
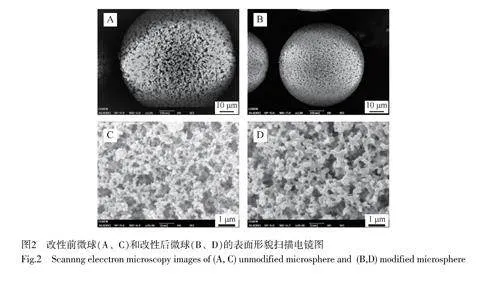
層析介質的結構對其色譜性能有重要影響。對制得的強陽離子交換層析介質的表面形貌進行觀察。采用掃描電子顯微鏡(SEM)觀察了改性前微球(Ⅰ)和改性后微球(Ⅳ)的形貌,如圖2所示。微球經改性后,骨架尺寸明顯變粗,表明烯丙基活化反應影響孔道內部結構。烯丙基的密度直接影響生物大分子在微球內部的傳輸,微球表面形貌顯示,經過改性后得到的介質仍然具有豐富的大孔結構,孔徑可達到1000nm,為實現蛋白快速傳質奠定了基礎[13]。
2.2活化微球烯丙基密度的影響因素考察
水解后的PGMA-EDMA微球表面富含羥基,利用羥基與烯丙基縮水甘油醚的環氧基進行反應,在微球表面引入烯丙基,此過程中伴隨AGE水解及交聯副反應的發生[14]。烯丙基密度對下一步加成生成磺酸基(SP)有直接影響,因此需要對其影響因素進行系統考察。
2.2.1AGE用量對烯丙基密度的影響
考察了不同AGE用量對微球烯丙基密度的影響。結果表明,隨著AGE用量增大,烯丙基密度不斷增大;當AGE用量達到2.67mL/g時,烯丙基密度為252.08μmol/g;繼續增加AGE用量,烯丙基密度基本保持不變。這主要是由于在反應初始階段微球表面含有大量羥基,隨著AGE不斷加入,烯丙基密度逐漸增大;隨反應持續進行,羥基含量不斷減少;繼續增加AGE用量,烯丙基密度不再發生明顯變化[15]。為了保證反應完全,選擇AGE的最佳用量為3mL/g。
2.2.2反應時間對烯丙基密度的影響
在3~18h反應時間范圍內,烯丙基密度呈上升趨勢;繼續延長反應時間,烯丙基密度基本保持不變,與文獻[16]報道的規律相似。這是因為在活化過程中,微球表面的羥基不斷被消耗,直至微球表面羥基完全被消耗,繼續延長反應時間,烯丙基密度基本不再發生變化。因此,選擇最佳反應時間為18h。
2.2.3反應溫度對烯丙基密度的影響
在30~70℃范圍內,隨著溫度升高,烯丙基密度也隨之增大;當溫度超過70℃后,繼續升高溫度,烯丙基密度反而下降。這主要是因為升高反應溫度可以加快羥基與環氧基的偶聯反應,但AGE的水解反應也隨之增加。因此,當溫度超過70℃時,烯丙基密度隨溫度升高而減小[17]。
2.2.4NaOH溶液濃度對烯丙基密度的影響
當NaOH溶液濃度由10%(m/V)增至40%(m/V)時,烯丙基密度由97.86μmol/g增至132.74μmol/g,變化較小;但是,當NaOH溶液濃度達到50%時,烯丙基密度迅速增大至266.83μmol/g;繼續增大NaOH溶液濃度,烯丙基密度增加不明顯。出現這種變化趨勢的原因是NaOH作為反應的催化劑[18],當其濃度增大到一定值時,極大地促進了微球基質表面羥基不斷轉變成氧負離子,不斷地進攻環氧基團位阻小的碳,進而導致環氧基開環,生成帶有烯丙基的產物。當NaOH溶液濃度高于50%(m/V)時,微球表面剩余的反應基團較少,因此,增大NaOH溶液質量分數并不會明顯增加烯丙基的密度。
2.3活化微球偶聯SP的影響因素考察
2.3.1Na2S2O3濃度對配基密度的影響
實驗結果表明,隨著Na2S2O3濃度增大,介質配基密度先增大后趨于平緩;當Na2S2O3濃度為1.5mol/L時,配基密度達到最大(0.377mol/L)。通過調節Na2S2O3的濃度,可以制備具有不同配基密度的強陽離子交換介質。本研究選擇Na2S2O3的最佳濃度為1.5mol/L。
2.3.2溶液pH對配基密度的影響
在其它反應條件相同的情況下,隨溶液pH值升高,配基密度先增大后減小,在pH=4.5時微球表面SP密度達到最大值(0.34mol/L)。這主要是由于在弱酸環境下,雙鍵與偏重亞硫酸鈉加成生成的SP與水再次加成生成醇[19],升高pH值會促進2種加成反應同時加快。當反應體系的pHgt;4.5時,隨著pH升高,雙鍵在副反應中消耗增多,用于主反應的雙鍵減少,最終使得配基密度逐漸降低。
2.3.3反應溫度對SP密度的影響
在25~50℃反應溫度范圍內,SP密度隨溫度升高呈先增大后減小的趨勢。當溫度低于35℃時,升高溫度有利于提高偶聯的SP密度。這是由于升溫使得主反應(生成SP反應)的反應速率大于副反應(烯丙基雙鍵的聚合和與水的加成反應)的反應速率。當反應溫度高于35℃時,不僅提高了加成生成SP的反應速率,而且由于生成SP的反應過程中不斷產生酸,使得雙鍵與水的加成反應的速率增大,導致反應體系中雙鍵數量減少,致使配基密度下降。因此,選擇最佳反應溫度為35℃。
2.3.4反應時間對SP密度的影響
隨著反應時間延長,配基密度呈現先增大后趨于穩定的趨勢。這是由于隨著偶聯反應不斷進行,微球表面的烯丙基不斷被消耗;當反應時間達到24h后,反應達到平衡,配基密度基本不再發生變化。最終確定最佳反應時間為24h。
2.4陽離子交換介質吸附性能的評價
配基是固定相表面連接的功能基團,是固定相與待分離物質相互作用的位點[20]。介質偶聯配基的密度對于蛋白吸附容量和蛋白擴散具有重要作用,配基密度越大,與蛋白質發生作用的活性位點越多,越有利于蛋白質的擴散和吸附。但是,隨著配基密度增大,空間位阻效應隨之增加[21-23]。配基密度還會影響蛋白回收率。因此,研究配基密度與介質吸附性能之間的關系具有重要意義。
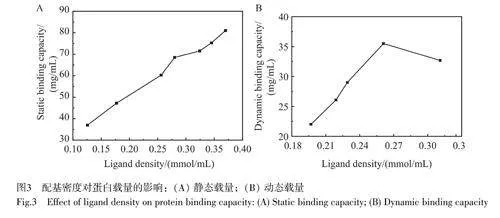
2.4.1配基密度對蛋白載量的影響
不同配基密度的陽離子交換介質的靜態吸附曲線見圖3A,在測定的配基密度范圍內,靜態載量隨配基密度增加而逐漸增大。這是由于隨著SP密度增加,介質表面與蛋白質的結合位點增多[24]。如圖3B所示,在配基密度低于0.261mmol/mL時,隨著配基密度增大,介質的動態載量逐漸增加;當配基密度大于0.261mmol/mL時,動態載量略有下降。這是由于配基密度較高時,孔道局部蛋白吸附量較多,部分孔道堵塞,導致蛋白傳質效率下降,最終導致蛋白動態吸附載量下降[25-26]。
2.4.2配基密度對溶菌酶回收率的影響
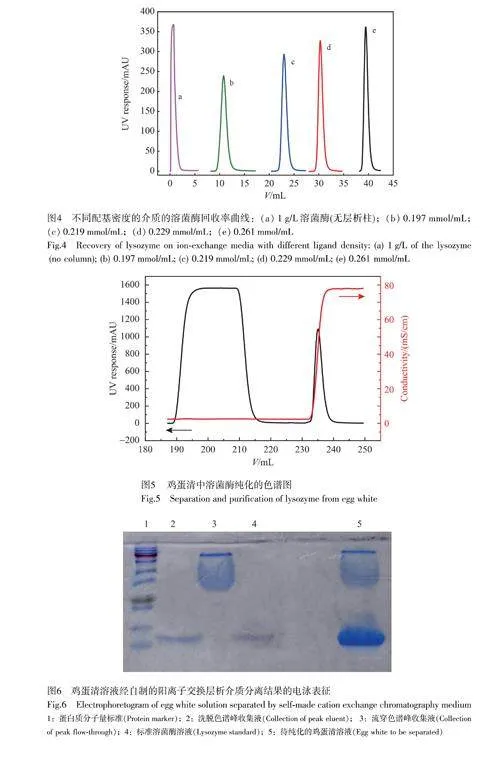
不同配基密度的陽離子交換介質對溶菌酶回收率的影響如圖4所示,曲線a代表空載上樣的溶菌酶色譜峰,曲線b~e依次是配基密度分別為為0.197、0.219、0.229和0.261mmol/mL的介質洗脫色譜峰。由圖4曲線b~e可見,隨著配基密度增加,溶菌酶的色譜峰面積逐漸增大,表明介質對溶菌酶的回收率增大,計算得到4種介質的回收率分別為95.85%、97.84%、98.77%和98.97%。這表明SP密度提高不僅增加了微球對溶菌酶的吸附位點,而且在一定程度上降低了微球的非特異性吸附。
2.5雞蛋清中溶菌酶的分離純化
采用自制的介質對雞蛋清中的溶菌酶進行分離,結果如圖5所示,色譜圖中只有1個流穿峰和1個洗脫峰。這是由于雞蛋清中含有的蛋白主要有卵蛋白(pI4.0)、卵清蛋白(pI4.6)、球蛋白(pI5.5~5.8)、伴清蛋白(pI6.6)和溶菌酶(pI10.8),當緩沖液pH=8.0時,雞蛋清中的蛋白除溶菌酶外,其余蛋白表面均帶有負電荷,不能被陽離子交換介質吸附,所以這些蛋白均流穿,此時只有帶有正電荷的溶菌酶被保留在介質上[27]。
分別對流穿液和洗脫液進行收集,通過凝膠電泳對其純度進行分析,結果如圖6所示,其中,泳道2為洗脫色譜峰收集液,泳道3為流穿色譜峰收集液,泳道4為標準溶菌酶溶液。由電泳圖可見,流穿液中不含溶菌酶,并且洗脫液中除溶菌酶外不含其它雜蛋白,證明制備的介質可以很好地實現溶菌酶的分離。
3結論
以大孔PGMA-EDMA微球為基質,通過水解、活化和偶聯三步反應制備了強陽離子交換層析介質。首先對烯丙基活化反應進行了系統考察,建立了烯丙基活化最優方法。基于此活化基質,系統考察了各因素對磺酸基偶聯反應的影響規律,獲得了偶聯磺酸配基的最佳制備工藝條件。對不同配基密度介質的吸附性能進行了比較,發現其蛋白結合載量隨離子交換容量增加而增大,同時其回收率也隨之上升。將制備的強陽離子交換層析介質用于雞蛋清中溶菌酶的分離,獲得了較好的分離效果。
