病毒感染與宿主抗感染免疫之間“博弈”
——凋亡、壞死和焦亡分子機(jī)制
2024-02-01 15:21:22陳松彪劉飛飛余祖華張春杰程相朝
畜牧獸醫(yī)學(xué)報 2024年1期
陳松彪,劉飛飛,尚 珂,余祖華,何 雷,魏 穎,陳 建, 張春杰,程相朝*,丁 軻*
(1.洛陽市活載體生物材料與動物疫病防控重點(diǎn)實(shí)驗室,洛陽 471003;2.河南科技大學(xué)功能微生物與畜禽健康 實(shí)驗室,洛陽 471003;3.動物病原與生物安全教育部重點(diǎn)實(shí)驗室,鄭州 450000)
病毒感染首先與宿主細(xì)胞表面受體相結(jié)合進(jìn)入細(xì)胞,病毒感染細(xì)胞后迅速“占領(lǐng)”宿主領(lǐng)地并將其改裝成為病毒加工制造“工廠”,其目的就是在此處大量合成產(chǎn)生新的感染性病毒粒子[1]。宿主控制和消除病毒復(fù)制傳播的有效方法之一就是通過程序性細(xì)胞死亡消除感染細(xì)胞,盡管有報道稱病毒在復(fù)制過程中誘導(dǎo)應(yīng)激導(dǎo)致細(xì)胞發(fā)生死亡,有助于病毒在感染后期釋放子代病毒粒子。但細(xì)胞早期死亡也誘導(dǎo)宿主形成了強(qiáng)大抗病毒防御機(jī)制:(1)細(xì)胞死亡破壞了病毒自身加工制造“工廠”;(2)細(xì)胞死亡大量釋放胞內(nèi)危險相關(guān)分子模式(damage associated molecular patterns,DAMPs)來促進(jìn)宿主固有免疫反應(yīng);(3)細(xì)胞死亡促進(jìn)了樹突狀細(xì)胞對病毒抗原的攝取和呈遞,促進(jìn)宿主適應(yīng)性免疫進(jìn)程[2-3]。宿主抗病毒感染免疫三種重要的細(xì)胞死亡途徑分別是凋亡(apoptosis)、壞死(necroptosis)和焦亡(pyroptosis),不同死亡模式由不同的特異信號級聯(lián)分子驅(qū)動[4-5],這些細(xì)胞死亡途徑啟動不同細(xì)胞形態(tài)學(xué)的解體過程。
許多病毒感染會導(dǎo)致宿主細(xì)胞的細(xì)胞膜失去完整性,從而有利于合成子代病毒釋放,病毒持續(xù)性感染對正常細(xì)胞功能的損害最終會導(dǎo)致細(xì)胞死亡。比如腺病毒E1A蛋白,它通過誘導(dǎo)細(xì)胞周期DNA復(fù)制期和有絲分裂期調(diào)節(jié)劑關(guān)鍵酶pRb(視網(wǎng)膜母細(xì)胞瘤抑制蛋白)失活,迫使宿主細(xì)胞復(fù)制周期進(jìn)入DNA合成的S期(有絲分裂間期),以大量合成脫氧核苷酸來促進(jìn)新病毒基因組DNA生成,在此過程中也誘導(dǎo)p53激活來促使內(nèi)源性死亡受體通路的細(xì)胞凋亡[6];與此相反,在此過程中病毒所編碼的E1B-55K蛋白還能夠通過與p53結(jié)合,來阻斷其促凋亡活性[7]。越來越多的證據(jù)顯示,病毒誘導(dǎo)宿主細(xì)胞死亡蛋白和拮抗宿主細(xì)胞死亡蛋白活性存在時間/空間上存活-死亡之間相對平衡[8-9]。近年來研究也發(fā)現(xiàn)病毒和宿主之間通過不同“博弈”來進(jìn)行感染和免疫,其中細(xì)胞凋亡、壞死和焦亡在此過程中發(fā)揮了重要作用。因此,本論文主要綜述了細(xì)胞凋亡、細(xì)胞壞死和細(xì)胞焦亡在病毒感染與宿主抗感染免疫過程中的作用及調(diào)控機(jī)制,以期能為深入研究病毒致病機(jī)制以及開發(fā)新的抗病毒藥物提供參考。
1 病毒感染與宿主抗感染免疫“博弈”——細(xì)胞凋亡
1.1 細(xì)胞凋亡
細(xì)胞凋亡又稱“細(xì)胞程序性免疫沉默”[10],是由半胱氨酸天冬氨酸蛋白酶(cysteinyl aspartate specific proteinase,Caspase)家族成員介導(dǎo)去除有害或者異常細(xì)胞等而產(chǎn)生細(xì)胞自我調(diào)節(jié)過程,即主動程序性細(xì)胞死亡方式[11]。當(dāng)病毒進(jìn)入細(xì)胞后,宿主模式識別受體(pattern recognition receptors, PRRs)對其基因組感知引發(fā)細(xì)胞凋亡是對宿主發(fā)揮先天免疫反應(yīng)抗病毒感染重要組成部分,病毒基因組可以以多種形式被感知,包括病毒DNA(vDNA)、病毒RNA合成的cDNA(v-cDNA)和病毒單鏈或雙鏈RNA(vRNA)。細(xì)胞凋亡受級聯(lián)反應(yīng)信號控制,并通過兩個重要信號通路發(fā)生:外源性的死亡受體通路(extrinsic death receptor pathway)和內(nèi)源性的線粒體通路(intrinsic mitochondrial pathway)[12-14]。
1.1.1 外源性死亡受體通路 外源性途徑是由細(xì)胞外的信號通過死亡配體如腫瘤壞死因子(tumor necrosis factor,TNF)、Fas配體(fas ligand,FasL)和腫瘤壞死因子相關(guān)的凋亡誘導(dǎo)配體(TNF-related apoptosis-inducing ligand,TRAIL)與各自的受體結(jié)合而啟動的[15]。死亡配體和相應(yīng)受體結(jié)合后募集Fas相關(guān)死亡結(jié)構(gòu)域(fas associated death domain,FADD)、Caspase-8前體(Pro-Caspase8)形成死亡誘導(dǎo)信號復(fù)合體(death inducing signaling complex,DISC),通過同型相互作用導(dǎo)致Caspase-8激活,激活的Caspase-8可直接水解激活效應(yīng)蛋白“凋亡執(zhí)行者”Caspase-7和Caspase-3誘導(dǎo)細(xì)胞凋亡[16](圖1)。
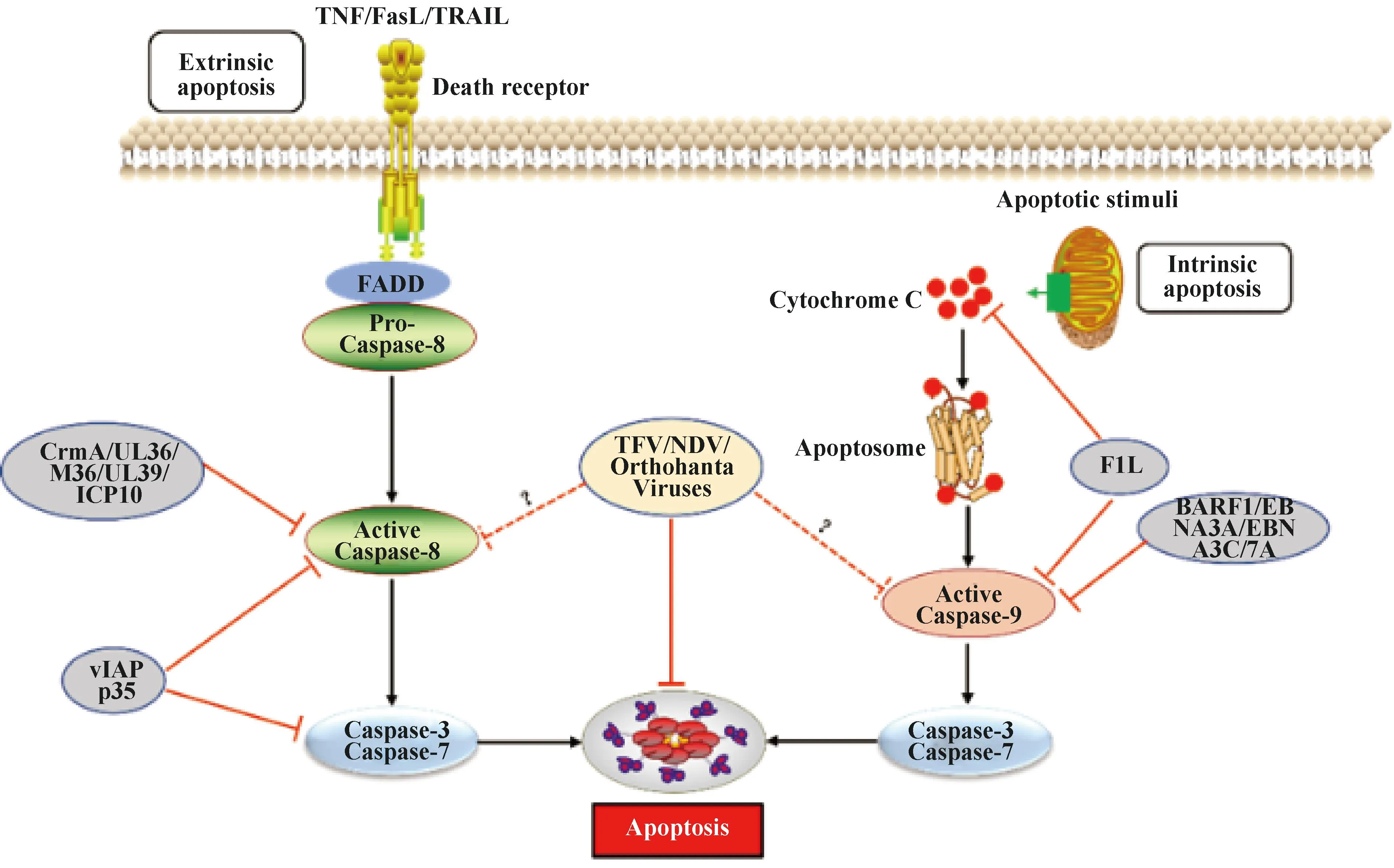
圖1 病毒感染與宿主抗感染免疫“博弈”——細(xì)胞凋亡Fig.1 Virus infection and host antiviral immunity struggle-cell apoptosis
1.1.2 內(nèi)源性線粒體通路 與外源性細(xì)胞凋亡不同,內(nèi)源性途徑是被DNA損傷、存活因子缺失、生長因子阻斷或其它細(xì)胞內(nèi)應(yīng)激等激活,引起B(yǎng)cl-2家族促凋亡蛋白(Bax、Bak、Bad、Bid、Puma、Bim和Noxa等)和抗凋亡蛋白(Bcl-2、Bcl-xl、Bcl-w和Mcl-1等)表達(dá)發(fā)生變化,然后促凋亡蛋白誘導(dǎo)線粒體外膜通透性(mitochondrial outer membrane permeabilization,MOMP)增加,從而導(dǎo)致細(xì)胞色素C(cytochrome C)從細(xì)胞器的膜間空間釋放,游離細(xì)胞色素C與Apaf-1結(jié)合形成凋亡小體復(fù)合物(apoptosome),該復(fù)合物進(jìn)一步激活Caspase-9來剪切下游的“凋亡執(zhí)行者”Caspases-3和Caspases-7誘導(dǎo)凋亡[17](圖1)。病毒感染可通過上調(diào)Bcl-2家族中促凋亡蛋白誘導(dǎo)細(xì)胞凋亡。人類免疫缺陷病毒Ⅰ型(HIV-1)感染能夠上調(diào)Puma和Bax水平[18-19];發(fā)熱伴血小板減少綜合征病毒[20]、哺乳動物呼腸孤病毒[21]、塔卡里伯病毒[22]以及丙型肝炎病毒[23]等感染上調(diào)Bak、Noxa或者Bik等促凋亡因子水平。同時,病毒感染也能夠下調(diào)Mcl-1或(和)Bcl-2等抗凋亡蛋白來誘導(dǎo)凋亡[24-25]。病毒感染誘導(dǎo)細(xì)胞凋亡表現(xiàn)為細(xì)胞有序解體和發(fā)生特定形態(tài)學(xué)變化[26],包括細(xì)胞皺縮、質(zhì)膜完整、細(xì)胞質(zhì)致密、細(xì)胞器密集、核裂解、細(xì)胞質(zhì)多發(fā)性芽突,并迅速脫落形成凋亡小體,這些凋亡小體被吞噬細(xì)胞吞噬后清除。
1.2 病毒感染抑制宿主細(xì)胞凋亡的分子機(jī)制
1.2.1 病毒感染抑制外源性死亡受體介導(dǎo)的細(xì)胞凋亡途徑 細(xì)胞凋亡是宿主抗病毒感染免疫的一種重要手段,病毒為了自身有效復(fù)制進(jìn)化出各種不同復(fù)雜機(jī)制來抵制宿主細(xì)胞凋亡。第一個由病毒自身編碼的凋亡抑制分子是牛痘病毒(Vaccinia virus,VACV)細(xì)胞因子修飾劑A(CrmA),CrmA具有廣泛的絲氨酸和半胱氨酸蛋白酶抑制活性,能夠有效抑制Caspase-1、Caspase-8和Caspase-9的活性[27],后續(xù)在其它痘病毒中也相繼發(fā)現(xiàn)CrmA的同源蛋白[28](圖1)。病毒凋亡抑制蛋白(vIAPs)是另一類廣譜Caspase抑制劑,vIAPs同源物在桿狀病毒感染的蝴蝶、飛蛾和蒼蠅中被發(fā)現(xiàn),大多數(shù)病毒和細(xì)胞IAPs包含具有泛素連接酶活性的桿狀病毒IAP重復(fù)序列和C末端的環(huán)指結(jié)構(gòu)域(really interesting new gene,RING),迄今為止發(fā)現(xiàn)超過200種vIAPs都是由感染昆蟲的大型DNA病毒所攜帶。多角體病毒vIAP的p35可作為人類Caspase-1、Caspase-3、Caspase-6、Caspase-7、Caspase-8和Caspase-10活化的抑制劑[29],進(jìn)一步結(jié)構(gòu)信息學(xué)分析發(fā)現(xiàn)p35可以和底物競爭性結(jié)合Caspase-8催化位點(diǎn)來抑制其活性,但是其如何抑制Caspase-1、Caspase-3、Caspase-6、Caspase-7和Caspase-10活性尚不清楚[30](圖1)。人巨細(xì)胞病毒(HCMV)、小鼠巨細(xì)胞病毒(MCMV)、單純皰疹病毒1型(HSV-1)以及HSV-2基因組編碼UL36[31]、M36[32]、UL39[33]以及ICP10[34]蛋白,能夠與Caspase-8相互作用,抑制其蛋白水解酶活性(圖1)。此外,γ-皰疹病毒和軟疣病毒屬成員表達(dá)c-FLIP(vFLP)同源物,一種類似于缺少Caspase-8催化活性蛋白酶結(jié)構(gòu)域的短c-FLIP亞型,c-FLIP與Caspase-8形成異質(zhì)二聚體,導(dǎo)致Caspase-8失活來阻斷其介導(dǎo)的細(xì)胞凋亡作用,從而阻斷凋亡信號通路[35-36]。
1.2.2 病毒感染抑制內(nèi)源性線粒體通路介導(dǎo)的細(xì)胞凋亡途徑 病毒感染后宿主可通過上調(diào)Bcl-2家族中促凋亡蛋白或下調(diào)抗凋亡蛋白誘導(dǎo)細(xì)胞凋亡,從而阻止病毒的復(fù)制[37]。但是,病毒在長期進(jìn)化過程中利用表達(dá)自身病毒蛋白也可以通過相應(yīng)策略來抑制凋亡[38]。EB病毒(Epstein-Barr virus,EBV)編碼的BARF1蛋白通過上調(diào)病毒感染細(xì)胞中抗凋亡蛋白Bcl-2和Bcl-xl的表達(dá)水平來抑制細(xì)胞凋亡[39],同時其編碼的EBNA3A和EBNA3C蛋白可下調(diào)促凋亡蛋白Bim的表達(dá)水平,抑制細(xì)胞凋亡[40](圖1)。乙型肝炎病毒(HBV)上調(diào)非編碼RNA lnc-HUR1來抑制p53轉(zhuǎn)錄活性,進(jìn)而上調(diào)凋亡抑制因子Bcl-2、抑制凋亡促進(jìn)因子Bax轉(zhuǎn)錄來抑制細(xì)胞凋亡[41]。此外,VACV、嚴(yán)重急性呼吸綜合征冠狀病毒(SARS-CoV)以及風(fēng)疹病毒(RV)等病毒可編碼蛋白通過與Bcl-2家族蛋白相互作用影響其正常功能抑制細(xì)胞凋亡[42-44]。VACV編碼F1L蛋白能夠結(jié)合Bcl-2家族蛋白,抑制細(xì)胞色素C釋放,從而抑制Caspase-9的激活。進(jìn)一步研究顯示F1L也可直接利用其N端結(jié)構(gòu)域與Caspase-9互作來抑制其活性[43],從而抑制由過表達(dá)Caspase-9所誘導(dǎo)凋亡[44](圖1)。RV衣殼蛋白通過直接與Bax結(jié)合抑制細(xì)胞凋亡,防止Bax激活誘導(dǎo)細(xì)胞凋亡[45]。對輪狀病毒屬[46]、副黏病毒屬[47]和布尼病毒屬[48]等成員研究也發(fā)現(xiàn)病毒感染過程中采取不同策略抑制細(xì)胞凋亡從而促進(jìn)自身復(fù)制,但具體作用機(jī)制尚不清楚。
2 病毒感染和宿主抗感染免疫“博弈”——細(xì)胞壞死
2.1 細(xì)胞壞死
由于外界不利因素引起細(xì)胞正常代謝活動受損或者發(fā)生中斷時,在受體相互作用蛋白激酶1(receptor-interacting protein kinase 1,RIPK1)及其下游的RIPK3和混合譜系激酶域樣蛋白(mixed lineage kinase domain-like,MLKL)將會激活細(xì)胞程序性壞死通路。細(xì)胞程序性壞死是一種新型的細(xì)胞炎性死亡方式,以胞質(zhì)內(nèi)蛋白形成壞死小體,細(xì)胞膜打孔破裂釋放內(nèi)容物誘導(dǎo)炎癥為主要特征。不同于細(xì)胞凋亡,壞死和焦亡都不參與維持細(xì)胞正常生理機(jī)能,而是以溶解和炎癥反應(yīng)為特征細(xì)胞死亡方式,具有重要抗病毒功能[49]。
細(xì)胞表面腫瘤壞死因子(tumor necrosis factor α,TNFα)受體1((tumor necrosis factor receptor 1,TNFR1)是死亡受體超家族的一員,其刺激可激活程序性壞死通路[49],TNFα與TNFR1結(jié)合形成由TNFR1相關(guān)的死亡結(jié)構(gòu)域蛋白 (TNFR1-associated death domain protein,TRADD)、TNF受體2(TNF receptor associated factor 2,TRAF2)、細(xì)胞內(nèi)凋亡蛋白質(zhì)抑制蛋白1/2(cIAP1/2)和RIPK1組成的膜信號復(fù)合物I(Complex I),隨后激活NF-κB通路從而促進(jìn)細(xì)胞存活和炎癥形成(圖2)。當(dāng)RIPK1泛素化被抑制時Complex I將被釋放并募集FADD,形成TRADD-FADD-Caspase-8組成復(fù)合物II(Complex II α),進(jìn)而激活凋亡信號通路。在病毒感染期間或?qū)aspase-8活性進(jìn)行藥物抑制,RIPK1和RIPK3借助它們的RIP同源基序互作結(jié)構(gòu)域(RIP homotypic interaction motif,RHIM)發(fā)生結(jié)合形成壞死小體,壞死小體啟動RIPK3介導(dǎo)的MLKL磷酸化,磷酸化的MLKL破壞質(zhì)膜并導(dǎo)致細(xì)胞壞死[50](圖2)。值得注意的是,在大多數(shù)生理條件下,RIPK1-RIPK3的細(xì)胞壞死復(fù)合體形成會受到cFLIP/Caspase-8異質(zhì)二聚體復(fù)合物抑制,該復(fù)合物通過接頭蛋白FADD被募集到RIPK1-RIPK3復(fù)合物中,并能同時降解RIPK1和RIPK3以防止壞死和促進(jìn)凋亡[51]。因此,Caspase-8活性的抑制(類似于MCMV表達(dá)vICA蛋白功能)是誘導(dǎo)細(xì)胞壞死的關(guān)鍵步驟。
近年來研究發(fā)現(xiàn),細(xì)胞壞死也可以被胞內(nèi)感受器Z-DNA結(jié)合蛋白1(ZBP1)激活,當(dāng)病毒釋放了核酸進(jìn)入細(xì)胞內(nèi),ZBP1能夠識別并結(jié)合細(xì)胞質(zhì)中的雙鏈DNA分子,同時激活RIPK3,使其磷酸化,獲得激酶活性,激活的RIPK3磷酸化MLKL,誘導(dǎo)細(xì)胞壞死(圖2)。在流感病毒和傳染性法氏囊病毒復(fù)制過程中產(chǎn)生Z-RNA(一種新的PAMP分子),這些Z-RNA激活感染核中的ZBP1,ZBP1被激活后刺激RIPK3,RIPK3發(fā)生磷酸化并激活細(xì)胞核中的MLKL,MLKL引發(fā)核膜破壞,促進(jìn)細(xì)胞DNA釋放至細(xì)胞質(zhì)中,從而誘導(dǎo)細(xì)胞壞死(圖2)[52-53]。
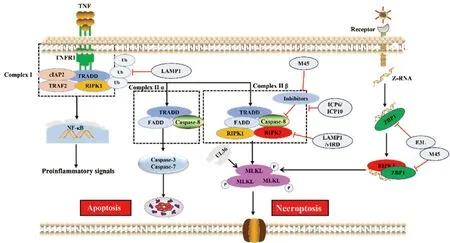
圖2 病毒感染與宿主抗感染免疫“博弈”——細(xì)胞壞死(參照文獻(xiàn)[53]并進(jìn)行修改)Fig.2 Virus infection and host antiviral immunity struggle-cell necroptosis (Modified according to reference[53])
2.2 病毒感染抑制宿主細(xì)胞壞死的分子機(jī)制
病毒針對細(xì)胞這一特性,進(jìn)化出各種復(fù)雜“武器”阻止細(xì)胞壞死這一自殺性行為,從而實(shí)現(xiàn)復(fù)制和生存。主要靶向于細(xì)胞壞死通路中的關(guān)鍵分子MLKL、RIPK1/RIPK3和ZBP1。
2.2.1 靶向于MLKL抑制細(xì)胞壞死 皰疹病毒家族多個成員編碼含有RHIM結(jié)構(gòu)域蛋白,干擾宿主細(xì)胞RIPK3組裝以及伴隨的下游MLKL激活。MCMV、HSV-1、HSV-2基因組編碼M45[54]、ICP6[55]、ICP10[34]蛋白通過依賴性N末端RHIM結(jié)構(gòu)域與RIPK1、TRIF以及ZBP1互作,競爭性抑制Complex II β復(fù)合體形成,減弱MLKL磷酸化從而抑制細(xì)胞壞死(圖2)。MCMV編碼具有酶活性UL36蛋白,與MLKL互作通過蛋白酶體途徑促進(jìn)MLKL降解,從而抑制壞死[31]。痘苗病毒(vaccinia virus,VV)編碼vIRD蛋白能夠促進(jìn)RIPK3泛素化和蛋白酶體降解途徑來抑制下游MLKL磷酸化來抑制細(xì)胞壞死[8]。
2.2.2 靶向于RIPK1和RIPK3抑制細(xì)胞壞死 牛痘病毒、鼠痘病毒和天花病毒均能表達(dá)另一種具有降解RIPK3功能的病毒蛋白vIRD,在病毒感染過程中vIRD通過其N端重復(fù)序列與RIPK3結(jié)合,通過其C端F-box與細(xì)胞SKP1-CUL1復(fù)合體結(jié)合促進(jìn)RIPK3的K48型鏈泛素化和蛋白酶體介導(dǎo)的降解,抑制感染細(xì)胞發(fā)生壞死[56]。EBV表達(dá)LMP1通過兩個不同途徑抑制壞死:干擾RIPK1和RIPK3泛素化以及降低RIPK3表達(dá)[57]。在痘病毒中還發(fā)現(xiàn)病毒編碼的蛋白COTV157和BAV Rmi具有和MLKL同源的假激酶結(jié)構(gòu)域(缺乏活性激酶的大多數(shù)或所有催化基序),通過它們和RIPK3互作競爭性阻止MLKL結(jié)合和磷酸化,從而抑制壞死[58]。
2.2.3 靶向于ZBP1抑制細(xì)胞壞死 VACV所編碼病毒蛋白E3L,該蛋白包含兩個不同的雙鏈RNA(dsRNA)結(jié)合結(jié)構(gòu)域,這兩個結(jié)構(gòu)域都對毒性至關(guān)重要:E3L蛋白N-末端含有Z-核酸結(jié)合域(Zα),它專門與Z-RNA結(jié)合,C-末端包含一個典型的dsRNA結(jié)合結(jié)構(gòu)域,它與典型的α構(gòu)象中的dsRNA相互作用。Zα結(jié)構(gòu)域與ZBP1的功能結(jié)構(gòu)域相似,能夠隔離Z-RNA的配體,阻止ZBP1的活化來抑制細(xì)胞壞死從而逃避宿主天然免疫反應(yīng)[59]。此外,MCMV病毒蛋白M45含有RHIM結(jié)構(gòu)域,可以干擾ZBP1和RIPK3之間的相互作用抑制細(xì)胞壞死(圖2)[60]。以上報道表明壞死不僅發(fā)生在Caspase-8活性被阻斷的情況下,而且可能作為一種單獨(dú)的機(jī)制來抑制病毒感染,但是凋亡-壞死之間切換信號及相應(yīng)調(diào)控機(jī)制仍不清楚。
3 病毒感染和宿主抗感染免疫“博弈”——細(xì)胞焦亡
3.1 細(xì)胞焦亡
細(xì)胞焦亡是細(xì)胞感染時由炎癥小體介導(dǎo),以裂解細(xì)胞為特點(diǎn)的程序性死亡形式,是機(jī)體重要的天然免疫反應(yīng),在拮抗和清除病原菌感染中發(fā)揮關(guān)鍵作用[61-62]。細(xì)胞焦亡激活信號途徑分為依賴Caspase-1的經(jīng)典途徑和依賴Caspase-4、Caspase-5或Caspase-11的非經(jīng)典途徑,這兩種途徑均通過切割炎性半胱氨酸蛋白酶D(gasdermin D,GSDMD),釋放出N端游離的肽段,該肽段會誘使細(xì)胞形成孔道導(dǎo)致細(xì)胞破裂發(fā)生焦亡。
3.1.1 依賴Caspase-1的信號通路 依賴Caspase-1的細(xì)胞焦亡通路又稱為經(jīng)典炎癥小體信號通路。炎癥小體是天然免疫系統(tǒng)的重要組成部分,能夠識別并激活病原菌/微生物相關(guān)分子模式(pathogen-associated molecular patterns,PAMPs/DAMPs),招募和激活促炎癥蛋白酶Caspase-1,其中最常見的炎癥小體是NOD樣受體蛋白(NLR family pyrin domain containing 1,NLRP1、NLRP3和NLRC4)和黑色素瘤缺乏因子2(absent in melanoma 2,AIM2)樣受體[4]。炎癥小體是一個多蛋白復(fù)合物,其激活通常是由PRRs與含有半胱天冬酶募集結(jié)構(gòu)域(caspase activation and recruitment domain,CARD)的凋亡相關(guān)斑點(diǎn)樣蛋白(apoptosis-associated speck-like protein containing a caspase-recruitment domain,ASC)之間的同質(zhì)相互作用誘導(dǎo)引發(fā),ASC通過CARD-CARD相互作用募集并激活Caspase-1,Caspase-1活化后切割I(lǐng)L-1β和IL-18前體,形成成熟促炎細(xì)胞因子IL-1β和IL-18[63]。活化的Caspase-1也可以切割GSDMD的N端和C端結(jié)構(gòu)域的連接區(qū),釋放具有結(jié)合膜磷脂上膜打孔活性的N端結(jié)構(gòu)域,導(dǎo)致細(xì)胞質(zhì)膜上形成孔,隨后細(xì)胞膜破裂,釋放大量炎癥細(xì)胞因子和DAMPs,引發(fā)細(xì)胞焦亡[9](圖3)。
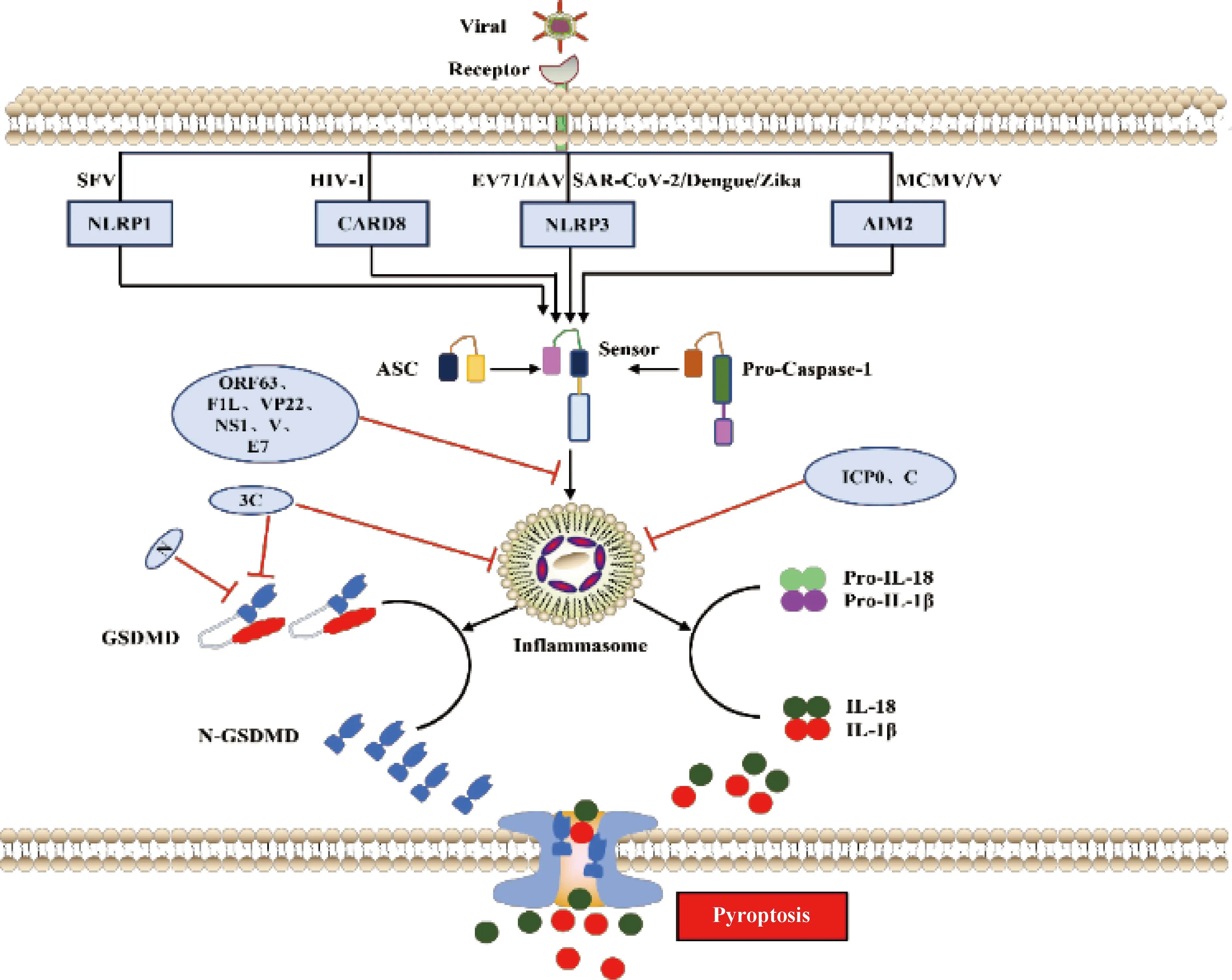
圖3 病毒感染與宿主抗感染免疫“博弈”——細(xì)胞焦亡(參照文獻(xiàn)[10]并加以修改)Fig.3 Virus infection and host antiviral immunity struggle-cell pyroptosis (Modified according to reference[10])
NLRP1在塞姆利基森林病毒(SFV)感染或合成的dsRNA模擬物poly(I:C)轉(zhuǎn)染過程中充當(dāng)dsRNA傳感器,NLRP1通過其富含亮氨酸的重復(fù)結(jié)構(gòu)域與dsRNA相互作用,誘導(dǎo)NLRP1寡聚以及激活Caspase-1形成炎癥小體,最終促使焦亡發(fā)生[64];登革病毒(Dengue virus)[65]、寨卡病毒(Zika virus)[66]、腸病毒(EV)[67]以及新型冠狀病毒(SARS-CoV-2)[68]感染后利用不同的手段促進(jìn)NLRP3表達(dá)發(fā)生上調(diào),促進(jìn)Capase-1的活化和炎癥小體形成來誘導(dǎo)焦亡;HIV-1感染利用CARD8主要作為HIV-1蛋白酶感受器,CARD8在其感染后誘導(dǎo)細(xì)胞焦亡過程中發(fā)揮重要作用[69];同時發(fā)現(xiàn)MCMV[70]和VV[71]感染后,通過AIM2識別并激活下游Caspase-1來誘導(dǎo)細(xì)胞焦亡。
3.1.2 依賴Caspase-4、Caspase-5或Caspase-11的信號通路 依賴Caspase-4、Caspase-5或Caspase-11信號通路又稱Caspase-1非依賴細(xì)胞通路或非經(jīng)典炎癥小體通路,該信號通路是由人源Caspase-4和Caspase-5或鼠源Caspase-11觸發(fā),這些Caspases活化后切割GSDMD(和Caspase-1相同切割位點(diǎn))并啟動細(xì)胞焦亡[72-73](圖3未顯示)。近些年對非經(jīng)典通路炎癥小體通路研究較少,下文主要闡述病毒抑制經(jīng)典通路之細(xì)胞焦亡。
3.2 病毒感染抑制宿主細(xì)胞焦亡的分子機(jī)制
雖然病毒感染過程中有許多炎癥小體和焦亡激活的例子,但病毒也可以通過調(diào)節(jié)炎癥小體激活或靶向焦亡“執(zhí)行者”GSDMD直接抑制細(xì)胞焦亡來促進(jìn)自身復(fù)制。
3.2.1 DNA病毒抑制宿主細(xì)胞焦亡 卡波齊肉瘤皰疹病毒編碼NLRP1同源物蛋白ORF63, ORF63可競爭性抑制NLRP1炎癥小體活化以及下游的Caspase-1介導(dǎo)的細(xì)胞焦亡[74]。VV基因組編碼vBcl2蛋白同源物F1L具有雙重功能,F1L體外結(jié)合并抑制NLRP1,缺乏F1L病毒感染巨噬細(xì)胞后導(dǎo)致Caspase-1激活和IL-1β分泌增加。缺失F1L造成病毒毒力在體內(nèi)減弱,引起發(fā)熱反應(yīng)改變和Caspase-1水解增加,同時伴隨感染小鼠肺部炎癥加速但不影響細(xì)胞死亡或病毒復(fù)制[75]。HSV-1可拮抗AIM2和IFI16介導(dǎo)的炎癥小體激活,HSV-1蛋白VP22與AIM2炎癥小體相互作用,阻止其寡聚化,從而抑制AIM2活化,而ICP0泛素連接酶活性則靶向IFI16,使其通過蛋白酶體途徑發(fā)生降解[76-77]。人乳頭瘤病毒E7蛋白通過促進(jìn)TRIM21介導(dǎo)的IFI16炎癥小體泛素化降解抑制細(xì)胞焦亡[78]。兔黏液瘤病毒和兔纖維瘤病毒表達(dá)病毒pyrin樣蛋白,這些蛋白與接頭蛋白ASC直接相互作用以抑制NLRP3炎癥小體組裝[79-80](圖3)。
3.2.2 RNA病毒抑制宿主細(xì)胞焦亡 除了DNA病毒抑制宿主細(xì)胞焦亡促進(jìn)自身復(fù)制外,一些RNA病毒也通過不同策略來抑制細(xì)胞焦亡。柯薩奇病毒和腦心肌炎病毒編碼3C蛋白酶可切割NLRP3和GSDMD使其失活,但NLRP1被相同的病毒蛋白酶裂解導(dǎo)致N-甘氨酸特異性降解,導(dǎo)致NLRP1活化[62,81]。人NLRP1和小鼠同源蛋白NLRP1B的病毒蛋白酶切割位點(diǎn)進(jìn)化成病毒多聚蛋白的切割位點(diǎn),這表明NLRP1蛋白與腸道病毒蛋白酶共同進(jìn)化來發(fā)揮宿主抗病毒感染[80]。甲型流感病毒的NS1以及副黏病毒家族成員麻疹病毒、仙臺病毒和尼帕病毒的V蛋白可阻斷NLRP3炎癥小體的激活[82-83](圖3)。人副流感病毒3型利用C蛋白通過蛋白酶體途徑降解NLRP3來阻止炎癥小體激活[84](圖3)。此外,SARS-CoV-2編碼N核衣殼蛋白通過阻止Caspase-1介導(dǎo)的切割和GSDMD的激活來阻斷人單核細(xì)胞焦亡和IL-1β分泌[85-86](圖3)。綜上所述,病毒感染宿主細(xì)胞后引發(fā)炎癥小體組裝激活是一個相當(dāng)復(fù)雜的生物學(xué)過程,宿主細(xì)胞通過細(xì)胞質(zhì)內(nèi)的感受器第一時間識別病毒的不同組分,如結(jié)構(gòu)蛋白和病毒核酸等,并產(chǎn)生相應(yīng)的炎癥小體進(jìn)而激活后續(xù)炎癥和免疫相關(guān)反應(yīng)導(dǎo)致細(xì)胞焦亡,從而抑制病毒感染和復(fù)制。同時,病毒可以利用合成自身編碼蛋白來抑制炎癥小體的組裝和激活,以逃避宿主細(xì)胞抗感染免疫。
4 小結(jié)與展望
病毒進(jìn)化出不同策略來拮抗感染過程中細(xì)胞凋亡、壞死和焦亡,從而促進(jìn)了自身復(fù)制。病毒成功逃避了宿主重要的抗感染免疫途徑——細(xì)胞死亡,可能會誘導(dǎo)出宿主細(xì)胞新的替代抗感染免疫途徑出現(xiàn)。同時當(dāng)細(xì)胞受到病原微生物感染刺激時,細(xì)胞是否由于處于不同生理狀態(tài)而導(dǎo)致一些細(xì)胞對病原微生物刺激“抵抗”從而發(fā)生凋亡、壞死或焦亡?這些細(xì)胞選擇性抗感染免疫信號和調(diào)控機(jī)制又是什么?
目前研究發(fā)現(xiàn)病毒感染和宿主抗感染免疫之間的博弈類型也存在著時間和空間上關(guān)聯(lián)。例如,豬細(xì)小病毒感染過程中能夠發(fā)生凋亡、自噬性死亡(完全自噬和不完全自噬的轉(zhuǎn)化)[87-90],在RNA病毒小反芻獸疫病毒感染過程中也發(fā)現(xiàn)存在第一波自噬流和第二波自噬流[91],然而這些不同通路之間互相切換的信號及其確切機(jī)制仍不清楚。另外,還有研究發(fā)現(xiàn)顆粒酶A和顆粒酶B能夠激活GSDMB和GSDME,從而引起腫瘤細(xì)胞死亡[92-93],那么以顆粒酶A/GSDMB和顆粒酶B/GSDME細(xì)胞死亡軸為基礎(chǔ)開發(fā)出小分子模擬物是否能夠臨床應(yīng)用清除腫瘤感染細(xì)胞?隨著標(biāo)簽蛋白和同位素示蹤在病毒學(xué)研究中廣泛應(yīng)用,使得人們能夠更清楚、更全面認(rèn)識在病毒感染不同時期細(xì)胞微環(huán)境所發(fā)生的不同類型變化。未來的研究可通過活細(xì)胞實(shí)時成像、病毒示蹤以及病毒反向遺傳操作技術(shù)綜合分析,來闡明病毒操縱宿主細(xì)胞死亡以及宿主抗感染免疫的分子機(jī)制,將為進(jìn)一步揭示病毒的致病機(jī)制和合理設(shè)計精準(zhǔn)靶點(diǎn)來選擇性干擾宿主細(xì)胞死亡的病毒拮抗劑提供新的思路。
