CRISPR/Cas9技術高效制備山羊SOCS2基因編輯胚胎
2024-02-01 15:28:12張晨儉李隱俠劉偉佳王慧利吳家順曹少先
畜牧獸醫學報 2024年1期
張晨儉,李隱俠,丁 強,劉偉佳,王慧利,何 南,吳家順,曹少先*
(1.南京農業大學動物科技學院,南京 210095;2.江蘇省農業科學院畜牧研究所,江蘇省畜禽精準育種工程研 究中心,農業農村部種養結合重點實驗室,南京 210014;3.山東農業大學動物科技學院,泰安 271017)
細胞因子信號傳導抑制因子2(suppressor of cytokine signaling 2,SOCS2),屬于細胞因子傳導抑制因子家族成員,是生長激素的負調控因子[1-3]。Metcalf等[4]研究發現,SOCS2基因敲除小鼠6周齡體重顯著增加,Starr等[5]研究表明,SOCS2能通過與GH競爭GHR的結合,負調控GH-IGF1通路抑制生長。山羊SOCS2基因包含3個外顯子,編碼198個氨基酸。SOCS2蛋白包括3個結構域,中心保守的SH2結構域、C末端SOCS盒和可變的N-端結構域[1]。SH2結構域識別受體(如GHR)或者配體的磷酸酪氨酸是SOCS2蛋白發揮功能的關鍵步驟[6-8],識別過程中SH2結構域中5個高度保守的氨基酸殘基(Arg73、Ser75、Ser76、Thr83和Arg96)通過氫鍵形成磷酸酪氨酸結合口袋,將磷酸化的酪氨酸殘基(如GHR的pY595)鎖定[9];N-端結構域包含一個擴展的 SH2子結構域 (ESS),連接SH2結構域和SOCS盒,使SH2結構域識別和捕獲的底物泛素化[10]。據報道,綿羊SOCS2的SH2結構域第96號精氨酸突變為半胱氨酸(p.R96C),使其無法與磷酸酪氨酸結合導致SOCS2蛋白活性喪失,與野生型相比p.R96C突變綿羊體重增加了18%[11]。
CRISPR/Cas9屬于第3代基因編輯技術,通過gRNA識別并引導核酸酶Cas9對靶基因進行切割產生雙鏈斷裂(DSB)[12],誘導DNA同源重組(HDR)[13-14]或非同源末端連接(NHEJ)修復[15],實現對靶基因編輯[16]。相較于前兩代基因編輯技術ZFN、TALENs,CRISPR/Cas9具有編輯效率高、操作簡便、成本低的優勢,自問世以來,已被廣泛應用于基因功能研究、疾病治療和動植物育種等領域[17-19]。但CRISPR/Cas9的編輯效率和特異性與sgRNA序列直接相關[20-21],設計篩選特異性好、編輯效率高的sgRNA對于制備基因編輯動物特別是大動物至關重要。
目前關于山羊SOCS2基因編輯的報道很少,僅見Zhou等[22]利用堿基編輯器編輯綿羊SOCS2基因p.R96C的報道,且編輯效率僅25.0%(1/4)。亟需在山羊胚胎水平開展高效精準sgRNA的設計、篩選,為高效制備SOCS2基因敲除山羊,培育快長型肉用山羊提供技術基礎。
1 材料與方法
1.1 試驗材料
106枚新鮮波雜山羊卵巢采集自江蘇省海安市屠宰場,浸泡于37 ℃生理鹽水中,通過保溫瓶帶回實驗室,進行卵母細胞分離和培養。
1.2 主要試劑
MEGAshortscriptTMT7 Transcription Kit、mMESSAGE mMACHINETMT7 ULTRA Transcription Kit、MEGAclearTMKit購于 Thermo fisher公司;QIAquick Gel Extraction Kit 購自Qiagen公司;GenCrispr Cas9 Nuclease 購于GenScript公司;pMDTM19-T Vector Cloning Kit、T4 DNA Ligase購自 TaKaRa 公司;Discover-sc Single Cell WGA Kit、DNA marker、2×Rapid Taq Master Mix、Phanta HS Super-Fidelity DNA Polymerase購自南京諾唯贊生物科技股份有限公司。
1.3 SOCS2 基因的sgRNA設計及載體構建
1.3.1 sgRNA引物設計 根據山羊SOCS2基因(XM_018047625)SH2的保守序列,使用在線網站CRISPOR(http://crispor.tefor.net/)設計山羊SOCS2基因的sgRNA靶序列,選擇Lindel評分[23]高的sgRNA,其長度為20 bp,PAM(protospacer adjacent motif)序列為-NGG。為方便sgRNA引物與載體連接,在sgRNA正義鏈5′端添加ACCG,反義鏈5′端添加AAAC,見表1。送北京擎科生物科技有限公司合成。
1.3.2 sgRNA表達載體構建 使用BsaⅠ核酸內切酶將pGL3-U6-sgRNA-PGK-puromycin質粒線性化后膠回收純化。分別將合成的sgRNA引物用超純水稀釋為100 mol·L-1,每對各取上下游5 μL混合,98 ℃變性10 min,室溫冷卻,在T4 DNA Ligase作用下與線性化質粒16 ℃連接過夜。轉化DH5α感受態細胞,挑取菌落進行PCR鑒定。上游引物為pGL3-sg-F:5′-CGATTAGTGAACGGATCTCGACG-3′,下游引物為sgRNA反義鏈,擴增體系為2×Rapid Taq Master Mix 12.5 μL,上、下游引物各1 μL,菌液1 μL,加去離子水至25 μL;擴增程序包括95 ℃預變性3 min;95 ℃變性15 s,52 ℃退火20 s,72 ℃延伸15 s,共35個循環;72 ℃延伸10 min。PCR產物經瓊脂糖凝膠電泳鑒定后選取陽性克隆送北京擎科生物科技有限公司測序。
1.4 sgRNA與Cas9 mRNA的體外轉錄
分別以質粒pX330和pGL3-U6-sgRNA-PGK-puromycin為模板,PCR擴增制備Cas9和sgRNA體外轉錄模板,將T7啟動子引入擴增引物(序列見表1)。使用Phanta HS Super-Fidelity DNA Polymerase高保真酶進行擴增。擴增程序為:98 ℃預變性3 min;98 ℃變性10 s,60 ℃退火30 s,72 ℃延伸(Cas9、sgRNA體外轉錄模板延伸時間分別為2.5 min、30 s),共35個循環;最后72 ℃延伸10 min。對擴增產物進行膠回收純化作為體外轉錄模板。Cas9 mRNA和sgRNA分別使用mMESSAGE mMACHINETMT7 ULTRA Transcription Kit和MEGAshortscriptTMT7 Transcription Kit進行體外轉錄,MEGAclearTMKit純化,通過Nano drop2000超微分光光度計檢測其濃度,瓊脂糖凝膠電泳檢測其完整性。將體外轉錄的sgRNA與Cas9 mRNA混合,調整其終濃度分別至50 ng·μL-1和100 ng·μL-1,置-80 ℃保存。
1.5 sgRNA/Cas9 蛋白體外切割活性測定
以山羊基因組序列為模板,使用Primer Premier 5在基因編輯靶區域上、下游500 bp附近設計檢測引物SOCS2-F、SOCS2-R,序列見表2。以山羊全基因組DNA為模板,參照“1.3.2”的方法,對基因編輯區域進行PCR擴增。PCR產物膠回收純化后進行體外切割。體系為:Cas9 蛋白5 U,sgRNA 0.2 μg,底物DNA 1 μg,10×Cas9 Buffer 5 μL,補充RNase free ddH2O至50 μL,對照組未加入sgRNA。37 ℃反應3 h后進行瓊脂糖凝膠電泳檢測。
1.6 卵母細胞體外成熟
參照Cripso等[24]的方法進行卵母細胞體外成熟培養。將收集到的卵丘-卵母細胞復合體(COCs)置于成熟培養基中,每孔500 μL成熟培養液中放入100個COCs并覆蓋石蠟油,在38.5 ℃、5% CO2、飽和濕度培養箱中體外成熟培養24 h。
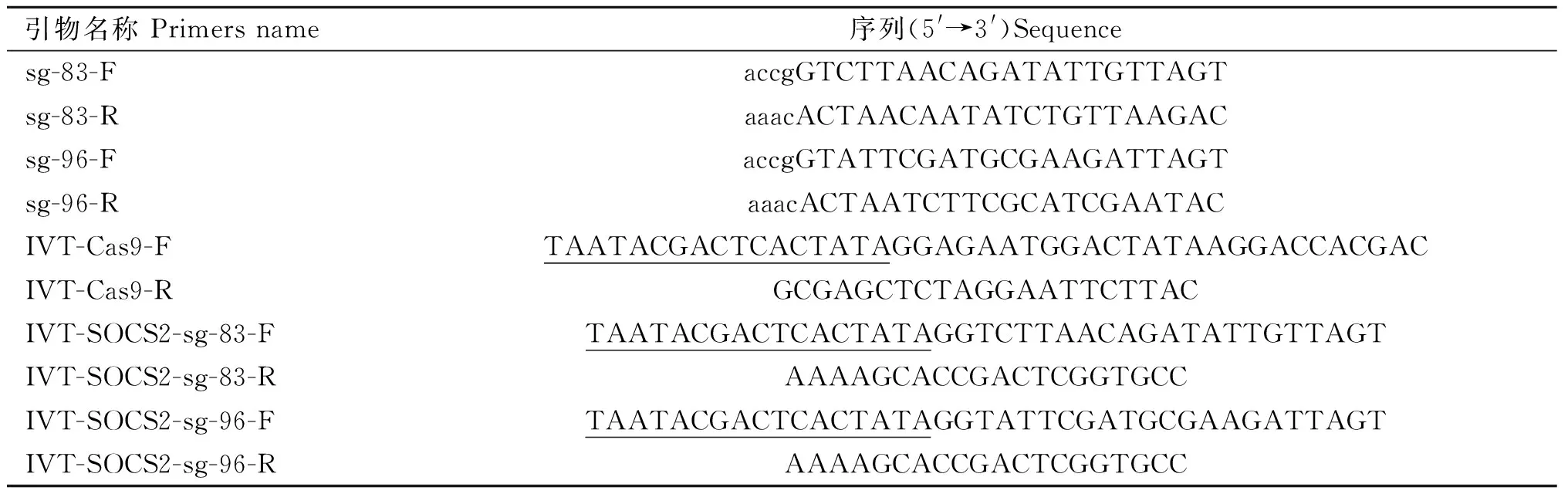
表1 sgRNA載體構建、Cas9和sgRNA體外轉錄模板制備引物Table 1 Oligonucleotides for constructing sgRNA vectors and making templates of Cas9 and sgRNA transcription in vitro
1.7 卵母細胞孤雌激活和顯微注射
參照Ledda等[25]的方法對MⅡ期卵母細胞進行孤雌激活,激活后繼續培養10~12 h,使用尼康顯微注射系統對單細胞孤雌胚胎注射Cas9 mRNA和sgRNA的混合物,對照組注射無核酸酶的超純水,繼續培養 7 d。激活后48 h統計卵裂率,第7 天收集囊胚。
1.8 基因編輯效率的檢測
收集單個囊胚于200 μL離心管中,按照Discover-sc Single Cell WGA Kit說明書進行單個胚胎全基因組DNA擴增。以擴增好的單個胚胎全基因組DNA 0.5~1 μL為模板,進行PCR擴增及測序鑒定,方法同“1.5”。在基因編輯靶區域附近出現雙峰的測序樣本,進一步進行克隆測序,每個樣本隨機選擇10個陽性菌落,分析編輯形式及效率。
1.9 脫靶效應預測及檢測
使用Cas-OFFinder(http://www.rgenome.net/cas-offinder/)在線網站對兩條sgRNA靶序列進行脫靶預測。每條sgRNA選擇5個錯配數最少的潛在脫靶位點,設計OFF-Target 檢測引物(見表2)進行PCR擴增測序,方法參照“1.3.2”。
2 結 果
2.1 山羊SOCS2基因sgRNA設計及載體構建
根據網站預測結果,如圖1所示,選擇SOCS2蛋白83號和96號氨基酸編碼密碼子附近的兩條sgRNA,靶序列分別為,SOCS2-sg-83:TCTTAACAGATATTGTTAGT,SOCS2-sg-96:TATTCGATGCGAAGATTAGT。采用菌落PCR鑒定sgRNA表達載體,如圖2所示,條帶大小符合預期,Sanger測序表明插入序列及方向正確,載體構建成功。
2.2 Cas9 mRNA和sgRNA的體外轉錄
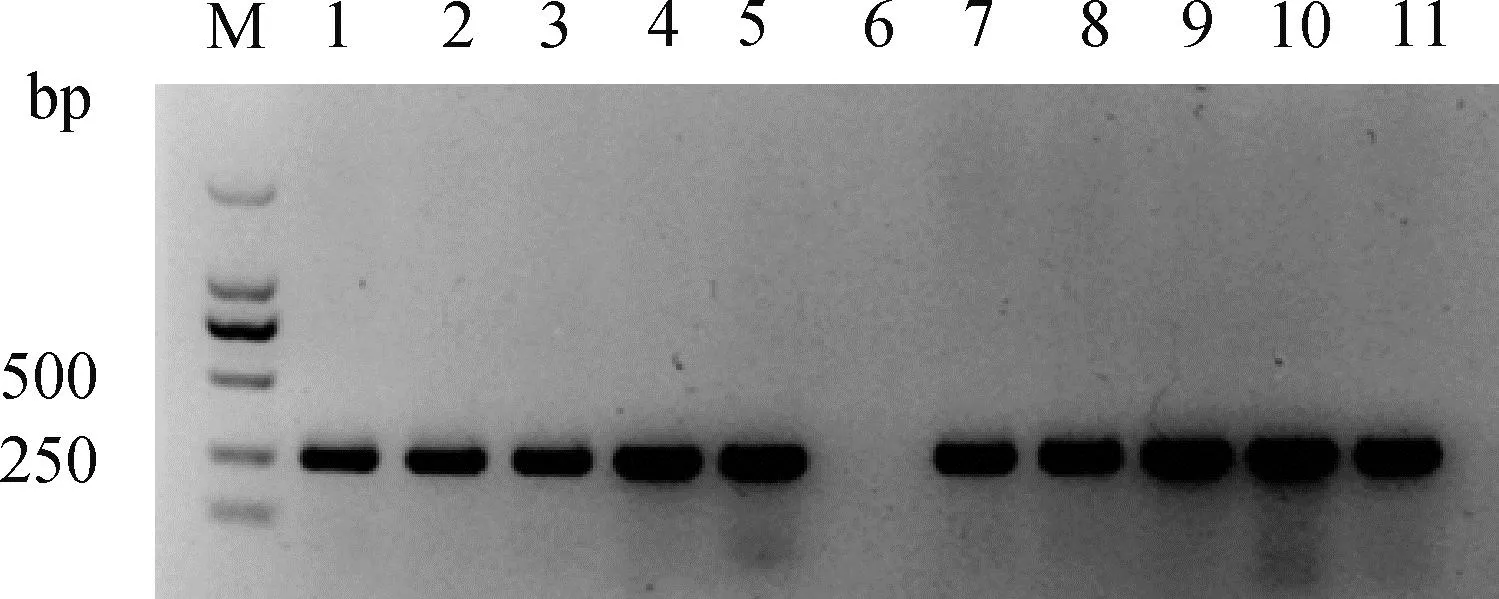
以pX330質粒和構建的sgRNA表達載體為模板,PCR制備Cas9和sgRNA體外轉錄模板,純化后的產物如圖3A、3B所示,條帶單一,大小符合預期;以此模板進行體外轉錄的產物如圖3C、3D所示,Cas9 mRNA和SOCS2-sg-83、SOCS2-sg-96主條帶明顯,片段大小符合預期,說明體外轉錄的mRNA質量較高。
2.3 sgRNA/Cas9蛋白體外切割活性檢測
如圖4A所示,對編輯區域進行PCR擴增,獲得了條帶單一,片段大小符合預期的切割底物,以此進行切割試驗,結果如圖4B所示,SOCS2-sg-83與SOCS2-sg-96兩位點均切割完全,在250 bp和500 bp之間出現兩條帶,SOCS2-sg-96位點兩條帶大小比較接近,與預期SOCS2-sg-83位點切割后條帶為287 bp和390 bp、SOCS2-sg-96位點切割后條帶為319 bp和358 bp相符,表明兩條sgRNA的體外切割效率高,可用于后續試驗。
2.4 顯微注射胚胎的體外發育
如表3所示,SOCS2-sg-83、SOCS2-sg-96分別注射胚胎152和140枚,48 h后卵裂率分別為75.7%和78.5%,囊胚率分別為55.7%和61.8%,與對照組無顯著差異。說明本試驗中的sgRNA和Cas9 mRNA未影響孤雌胚胎體外發育。

表2 基因編輯檢測引物及潛在脫靶位點引物Table 2 Primers of gene editing detection and potential off-target sites detection
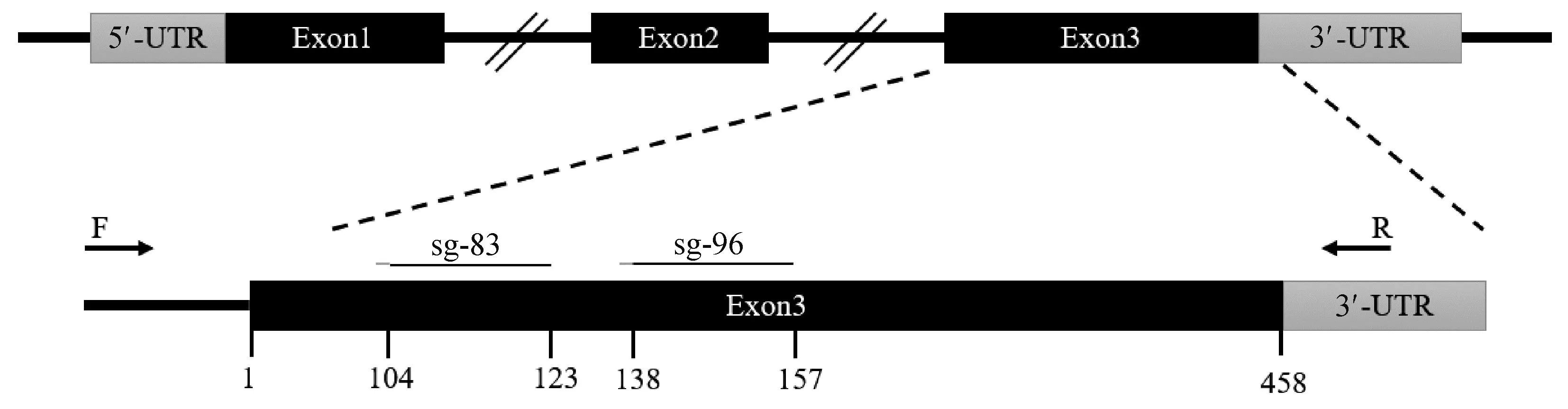
圖1 山羊SOCS2基因sgRNA靶位點示意圖Fig.1 Schematic diagram of the sgRNA target sites in the SOCS2 of goat
M. DNA相對分子質量標準;1~5、7~11. 陽性克隆;6. 陰性克隆 M. DL 2000 DNA marker; 1-5, 7-11. Positive colonies; 6. Negative colony圖2 sgRNA表達載體PCR鑒定Fig.2 Identification of sgRNA expression vectors by PCR
2.5 CRISPR/Cas9編輯胚胎SOCS2基因效果檢測
從64個注射SOCS2-sg-83的囊胚中隨機選取17個進行全基因組擴增和PCR檢測,如圖5A所示,產物大小符合預期,83-1樣本出現兩條帶,疑似出現大片段缺失。對PCR產物進行測序,其中有16個樣本在PAM區附近出現雙峰,如圖6所示,說明在此處發生了基因編輯,效率為94.1%;從68個注射SOCS2-sg-96的囊胚中隨機選取16個進行全基因組擴增和PCR檢測,如圖5B所示,產物條帶單一,大小符合預期,測序后發現8個樣本在PAM區附近出現雙峰,效率為50.0%,見圖6。同時,Cas9/sgRNA介導的突變發生在PAM區附近,符合Cas9切割的預期,進一步證明了該過程的靶向性。
A. Cas9體外轉錄模板制備:M. DNA相對分子質量標準;B. sgRNA 體外轉錄模板制備:M. DNA相對分子質量標準;C. Cas9 mRNA 體外轉錄:M. DNA相對分子質量標準;D. sgRNA 體外轉錄:M. DNA相對分子質量標準 A. Production transcription template of Cas9 in vitro: M. DL5000 DNA marker; B. Production transcription template of sgRNA in vitro: M. DL2000 DNA marker; C. Transcription of Cas9 mRNA in vitro: M. DL5000 DNA marker; D. Transcription of sgRNA in vitro: M. DL2000 DNA marker圖3 Cas9和sgRNA體外轉錄Fig.3 Cas9 and sgRNA in vitro transcription
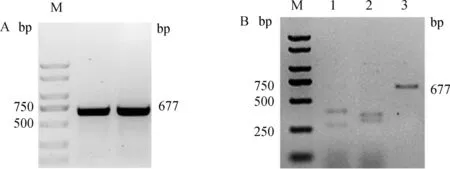
A. 切割底物PCR擴增;B. sgRNA/Cas9 體外切割活性測定:M. DL2000 DNA Marker;1. SOCS2-sg-83+Cas9;2. SOCS2-sg-96+Cas9;3. 對照組 A. Cleavage substrate PCR amplification; B. sgRNA/Cas9 cleavage activity detection in vitro: M. DL2000 DNA marker; 1. SOCS2-sg-83+Cas9;2. SOCS2-sg-96+Cas9;3. Control group圖4 sgRNA/Cas9體外切割活性的檢測Fig.4 Detection of sgRNA/Cas9 cleavage activity in vitro

表3 顯微注射的山羊孤雌胚胎發育情況Table 3 Development rate of goat parthenogenetic embryos after microinjection
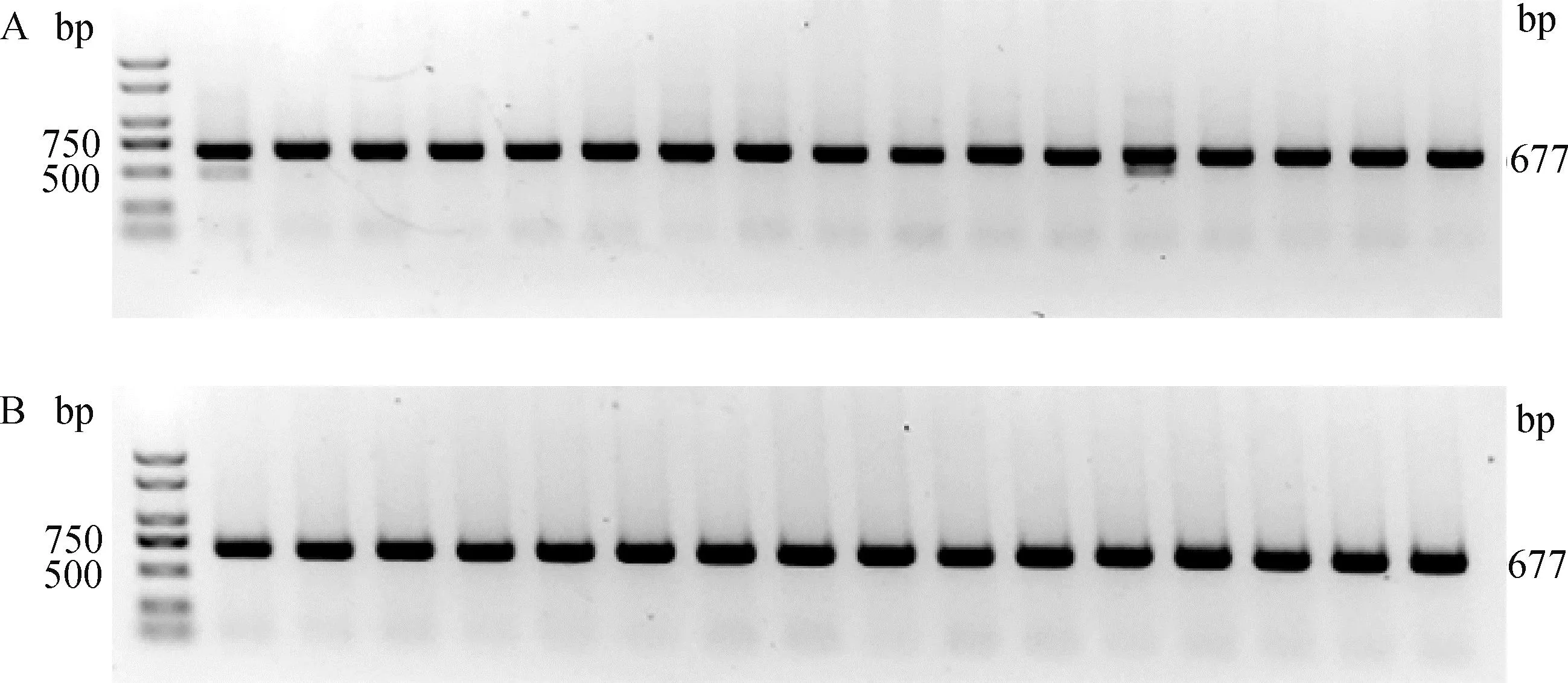
A. SOCS2-sg-83編輯囊胚PCR鑒定;B. SOCS2-sg-96編輯囊胚PCR鑒定 A. PCR identification of SOCS2-sg-83 edited blastocyst; B. PCR identification of SOCS2-sg-96 edited blastocyst圖5 囊胚中CRISPR/Cas9編輯SOCS2基因效果的PCR檢測Fig.5 PCR detection of the effect of CRISPR/Cas9 editing SOCS2 gene in blastocysts
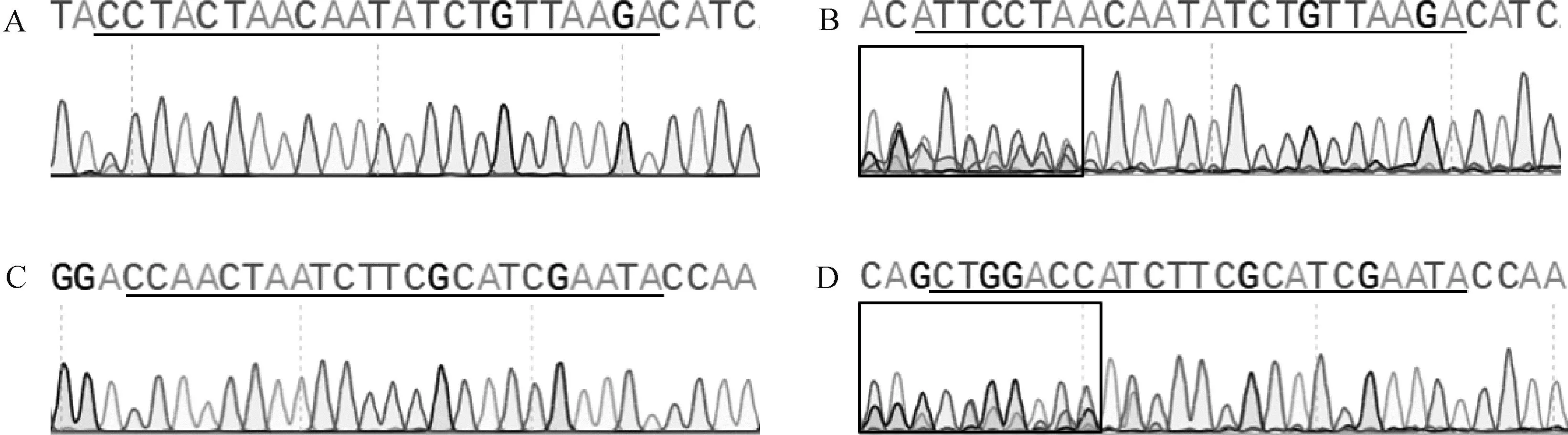
A. 對照組SOCS2-sg-83靶位點附近峰圖;B. 編輯組SOCS2-sg-83靶位點附近峰圖;C. 對照組SOCS2-sg-96靶位點附近峰圖;D. 編輯組SOCS2-sg-96靶位點附近峰圖 A. Sequencing peak near SOCS2-sg-83 target site in the control group; B. Sequencing peak near SOCS2-sg-83 target site in the edited group; C. Sequencing peak near SOCS2-sg-96 target site in the control group; D. Sequencing peak near SOCS2-sg-96 target site in the edited group圖6 囊胚中CRISPR/Cas9 編輯SOCS2基因效率的測序鑒定Fig.6 Sequencing identification of CRISPR/Cas9-edited SOCS2 gene efficiency in blastocysts
為了進一步研究CRISPR/Cas9在胚胎中的編輯形式,將PCR產物進行克隆測序,結果顯示(表4),在SOCS2-sg-83編輯的160個克隆中152個出現插入、缺失或替換,概率為95.0%,最多的突變類型為插入一個T(概率為33.8%);移碼突變(3N+1、3N+2)概率為81.3%(130/160)。此外,Indels為3N的克隆中有12個缺失83號氨基酸密碼子,可能破壞SH2結構域;有兩個插入了30個核苷酸,并引入了終止密碼子(TAA);另一個插入了36個核苷酸,也引入終止密碼子(TGA),將在翻譯過程中提前終止。因此,共有90.6%的克隆可能實現對SOCS2基因功能的敲除。SOCS2-sg-96編輯的80個克隆中70個出現插入、缺失或替換,概率為87.5%,最多的突變類型為缺失4 bp,概率為36.3%(29/80),移碼突變的概率為75.0%(60/80)。另外,在9個Indels為3N的克隆中,4個缺失第96號氨基酸密碼子。共計80.0%(64/80)的Indels可能實現對SOCS2基因功能的敲除(表5)。
2.6 脫靶效應分析
根據Cas-OFFinder在線網站預測SOCS2-sg-83、SOCS2-sg-96的脫靶位點,結果顯示,SOCS2-sg-83的預測位點中,錯配數為2的有1個,錯配數為3的有15個,錯配數為4的有143個;SOCS2-sg-96的預測位點中,錯配數為3的有2個;錯配數為4的有25個。每個sgRNA選擇5個錯配數最少的脫靶位點進行PCR擴增測序,靶序列如表6所示,其中小寫字母表示錯配堿基。測序結果顯示在選取的目標脫靶位點上未發現脫靶現象,如圖7、圖8所示。
3 討 論
SOCS2是細胞因子傳導抑制因子(SOCS)家族成員,通過其SH2結構域與GHR的磷酸化酪氨酸結合,阻斷GH與GHR的結合,發揮抑制生長的作用[26]。組成SH2磷酸酪氨酸結合口袋的5個氨基酸殘基(Arg73、Ser75、Ser76、Thr83和Arg96)在不同物種間高度保守,發生突變將影響其結合能力[9-10]。研究表明,SOCS2的p.R96C突變導致SH2與磷酸酪氨酸無法結合,SOCS2功能喪失[11]。本試驗選擇山羊SOCS2基因序列保守的83和96號氨基酸附近設計sgRNA,試圖在基因編輯造成移碼突變的同時也能通過破壞關鍵結構域導致基因的功能性敲除,提高基因敲除的效率,為高效制備SOCS2基因敲除山羊提供技術支撐。
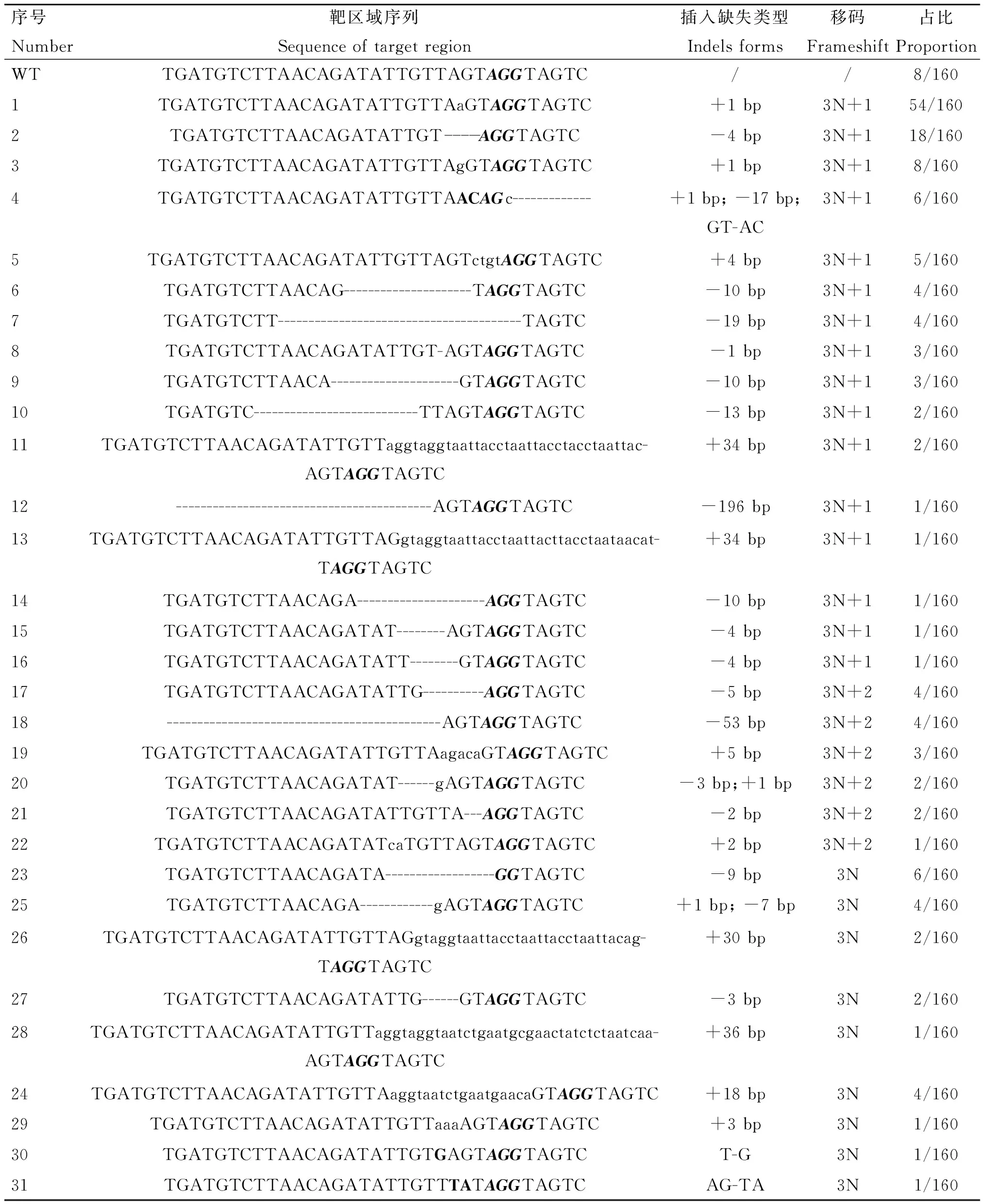
表4 SOCS2-sg-83不同編輯形式Table 4 Sequence varieties edited by SOCS2-sg-83
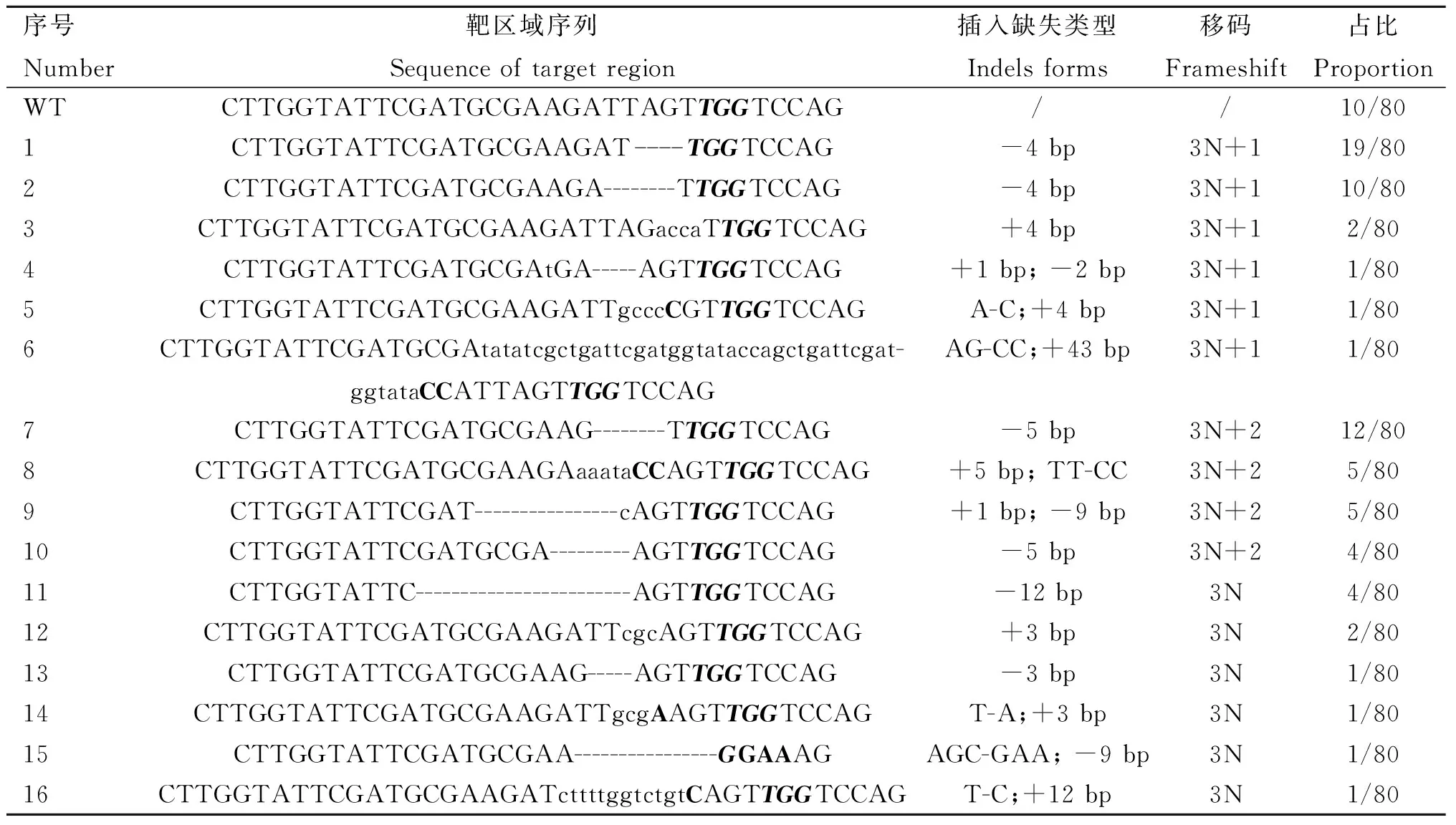
表5 SOCS2-sg-96不同編輯形式Table 5 Sequence varieties edited by SOCS2-sg-96

表6 sgRNA潛在脫靶位點預測Table 6 Potential off-target site prediction of sgRNA
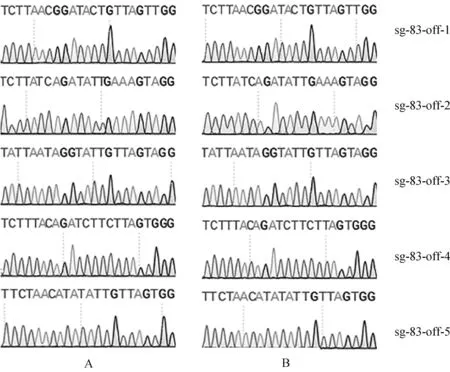
A.對照組;B.編輯組 A. Control group; B. Edited group圖7 SOCS2-sg-83脫靶序列測序分析Fig.7 The off-target analysis of SOCS2-sg-83 by sequencing
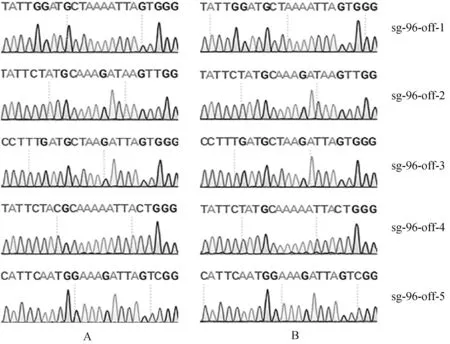
A. 對照組;B. 編輯組 A. Control group; B. Edited group圖8 SOCS2-sg-96 脫靶序列測序分析Fig.8 The off-target analysis of SOCS2-sg-96 by sequencing
盡管CRISPR/Cas9系統的基因編輯效率相較ZFN、TALENs等技術大幅提高,但許多研究中基因編輯效率仍在50.0%以下。如尹智等[27]對豬孤雌囊胚α-1,3-半乳糖苷轉移酶(GGTA1)基因的編輯效率為28.1%;尉翔棟等[28]編輯牛胚胎的肌肉生長抑素(MSTN)基因,編輯效率為15.4%;Zhang等[29]對綿羊孤雌胚胎的骨形態發生蛋白受體ⅠB(BMPR-ⅠB)基因進行編輯,效率為37.5%;目前,對SOCS2基因編輯的研究較少,僅見Zhou等[22]利用堿基編輯器對綿羊SOCS2的p.R96C位點進行編輯,效率為25.0%。這對于制備基因編輯動物是不利的,尤其是大型家畜的成本很高,有必要進一步提高編輯效率。研究表明,使用相同的基因編輯工具時,基因編輯的效率由sgRNA決定[30-32]。本試驗通過設計、篩選獲得的SOCS2-sg-83編輯胚胎的效率達到94.1%,可實現對胚胎的高效編輯。除了sgRNA的設計外,高質量轉錄的sgRNA和Cas9 mRNA也是本試驗高編輯效率的重要保障。
基因編輯主要通過移碼突變實現基因功能敲除[33-35],因為移碼突變會破壞蛋白質的編碼[36]。Zhang等[29]編輯綿羊胚胎BMPR-ⅠB基因,T-A克隆測序發現47.9%(78/163)的克隆存在移碼突變。Hu等[37]對綿羊成纖維細胞生長因子(FGF5)基因進行編輯,造成缺失5 bp的移碼突變,概率為66.7%(2/3),移碼突變破壞了蛋白質一級結構并提前引入終止密碼子,基因編輯羊的羊毛長度顯著長于野生型(P<0.05)。本試驗設計sgRNA時,選擇了網站移碼突變預測結果Lindel評分均為82的SOCS2-sg-83、SOCS2-sg-96,T-A克隆檢測結果顯示二者均可使SOCS2基因高效產生移碼突變,其中SOCS2-sg-83引發移碼突變的概率是81.3%,SOCS2-sg-96則為75.0%,高于66.7%的理論平均值。此外,在非移碼突變的情況下,破壞關鍵結構域的核心氨基酸將導致蛋白質功能失活,從而實現基因功能敲除。如綿羊SOCS2的SH2結構域第96號氨基酸由精氨酸突變為半胱氨酸(p.R96C)可導致SOCS2功能失活,本試驗中,SOCS2-sg-83引發的Indels為3 N的克隆中有7.5%(12/160)缺失83號氨基酸密碼子,可能破壞SH2結構域;另外,有1.3%(2/160)插入了30個核苷酸,并引入了終止密碼子(TAA),也可實現SOCS2基因功能性敲除。總之SOCS2-sg-83引發的突變中有90.6%可望實現SOCS2基因功能性敲除。此外,山羊SOCS2-sg-83在綿羊SOCS2基因SH2結構域中有完全匹配的靶序列,因此也可應用于綿羊SOCS2的基因編輯。
基因編輯中,sgRNA可能引導Cas9結合到與靶DNA相似的序列,產生非特異性編輯導致脫靶[38-40]。為此,本試驗對每條sgRNA選擇5個錯配數最少的潛在脫靶位點進行了檢測,未發現脫靶現象,說明SOCS2-sg-83、SOCS2-sg-96特異性較好,與郭日紅[41]、趙為民等[42]的研究結果類似。此外,本試驗中的sgRNA未影響孤雌胚胎發育,也從側面印證了sgRNA的特異性。
4 結 論
本研究利用CRIPSR/Cas9 系統在胚胎水平獲得了高效敲除山羊SOCS2基因的sg-SOCS2-sg-83,編輯效率高達94.1%,其中90.6%編輯可望造成SOCS2基因功能失活,且未發現脫靶,表明SOCS2-sg-83可實現山羊胚胎SOCS2基因的高效、精準編輯和敲除,為高效制備SOCS2基因敲除山羊,培育快長型肉用山羊奠定了技術基礎。
