催化劑中金屬與載體相互作用的表征研究進(jìn)展
2024-03-11 08:24:44彭潔李曜儲(chǔ)政于楊
天然氣化工—C1化學(xué)與化工 2024年2期
彭潔,李曜,儲(chǔ)政,于楊
(中石化南京化工研究院有限公司,江蘇 南京 210048)
早在1978年,相關(guān)人員在研究TiO2負(fù)載的貴金屬催化劑時(shí),提出了強(qiáng)金屬-載體相互作用(SMSⅠ)的概念[6-7]。之后,MSⅠ得到了更廣泛關(guān)注。MSⅠ普遍存在于負(fù)載型催化劑中,有多種表現(xiàn)形式,包括傳統(tǒng)經(jīng)典的SMSⅠ、氧化金屬-載體相互作用(OMSⅠ)、共價(jià)金屬載體相互作用(CMSⅠ)和電子金屬-載體相互作用(EMSⅠ)等[8-13],這些相互作用的具體表現(xiàn)形式由活性金屬和載體的類型和性質(zhì)決定。MSⅠ通常會(huì)導(dǎo)致載體對(duì)金屬的“包覆”、界面電荷轉(zhuǎn)移和催化劑對(duì)小分子的吸附減弱等現(xiàn)象,進(jìn)而影響催化劑的催化活性、選擇性及穩(wěn)定性[11,13]。
近年來(lái),隨著表征技術(shù)的不斷發(fā)展,特別是原位表征方法和高分辨電鏡技術(shù)的進(jìn)步,人們對(duì)負(fù)載型催化劑的認(rèn)識(shí)也不斷深入,從表觀的結(jié)構(gòu)特征到金屬-載體界面原子間的電荷轉(zhuǎn)移,從靜態(tài)物化性質(zhì)到實(shí)際反應(yīng)過(guò)程中的相互作用動(dòng)態(tài)變化,MSⅠ的表征研究已非常廣泛[11,14]。但是目前對(duì)于MSⅠ的報(bào)道多是對(duì)某一種或某幾種催化劑材料進(jìn)行針對(duì)性研究,相關(guān)綜述文章多是從MSⅠ的概念、形成、影響或應(yīng)用展開,雖然部分文章會(huì)列舉使用到的表征方法,但對(duì)MSⅠ的表征方法的系統(tǒng)性總結(jié)較少[10,14-15]。本文基于非原位和原位表征技術(shù),包括高分辨透射電子顯微鏡(HRTEM)、拉曼光譜(Raman)、X射線光電子能譜(XPS)和電子順磁共振譜(EPR)等,從MSⅠ的靜態(tài)性質(zhì)(如物質(zhì)輸送導(dǎo)致的微觀結(jié)構(gòu)特征、電荷轉(zhuǎn)移等)和動(dòng)態(tài)性質(zhì)(相互作用強(qiáng)弱變化即作用可逆性、界面結(jié)構(gòu)變化和電荷轉(zhuǎn)移過(guò)程等)展開,概述MSⅠ的表征研究進(jìn)展(圖1)。
1 MSⅠ的非原位表征
通過(guò)非原位手段,可以表征催化劑預(yù)處理后或反應(yīng)前后穩(wěn)定狀態(tài)下的MSⅠ。例如,HRTEM可以觀測(cè)金屬-載體界面的微觀結(jié)構(gòu),從而得到質(zhì)量轉(zhuǎn)移信息;紅外光譜(FTⅠR)、Raman等可以表征金屬-載體之間的成鍵情況;XPS、EPR等可以用于分析金屬-載體之間的界面電荷轉(zhuǎn)移。
1.1 MSI下的界面微觀結(jié)構(gòu)表征
SⅠNGH等[16]早在1985年便通過(guò)HRTEM觀測(cè)到了Rh/TiO2催化劑中Rh顆粒表面包覆的氧化物載體,這種“包覆”表明Rh與TiO2載體之間存在強(qiáng)相互作用,之后HRTEM逐漸成為表征金屬-載體界面結(jié)構(gòu)和質(zhì)量轉(zhuǎn)移的最常用方法[17-20]。早期人們認(rèn)為由于Au的功函和表面能較低,不易產(chǎn)生SMSⅠ,但近年來(lái),對(duì)于Au-載體之間相互作用的研究逐漸增多。經(jīng)過(guò)高溫焙燒之后,Au與載體之間可以產(chǎn)生不同程度的相互作用。TANG等[19]研究發(fā)現(xiàn),羥磷灰石(HAP)載體在Au納米顆粒表面的包裹程度取決于焙燒溫度(圖2)。由圖2可知,焙燒溫度為300 ℃時(shí),納米顆粒開始被薄層覆蓋,隨著溫度升高,覆蓋程度逐漸增大,600 ℃時(shí)Au納米顆粒被完全覆蓋。之后,Au/H-500-H2樣品(數(shù)字為焙燒溫度)在H2氛圍下還原,覆蓋層逐漸褪去。為了確認(rèn)覆蓋層的組分,還對(duì)Au/H-600樣品進(jìn)行了電子能量損失譜(EELS)分析,結(jié)果顯示覆蓋層信號(hào)與HAP載體相同,即表明在焙燒過(guò)程中載體遷移至Au納米顆粒表面,形成SMSⅠ。類似地,LⅠU等[20]通過(guò)HRTEM觀測(cè)到Au/ZnO催化劑經(jīng)過(guò)300 ℃氧化處理之后,ZnO納米棒載體組分向Au納米球遷移,包覆層的晶格間距為0.26 nm,與ZnO(002)晶面一致。H2氛圍下還原后,包覆層褪去,符合SMSⅠ的典型特征。
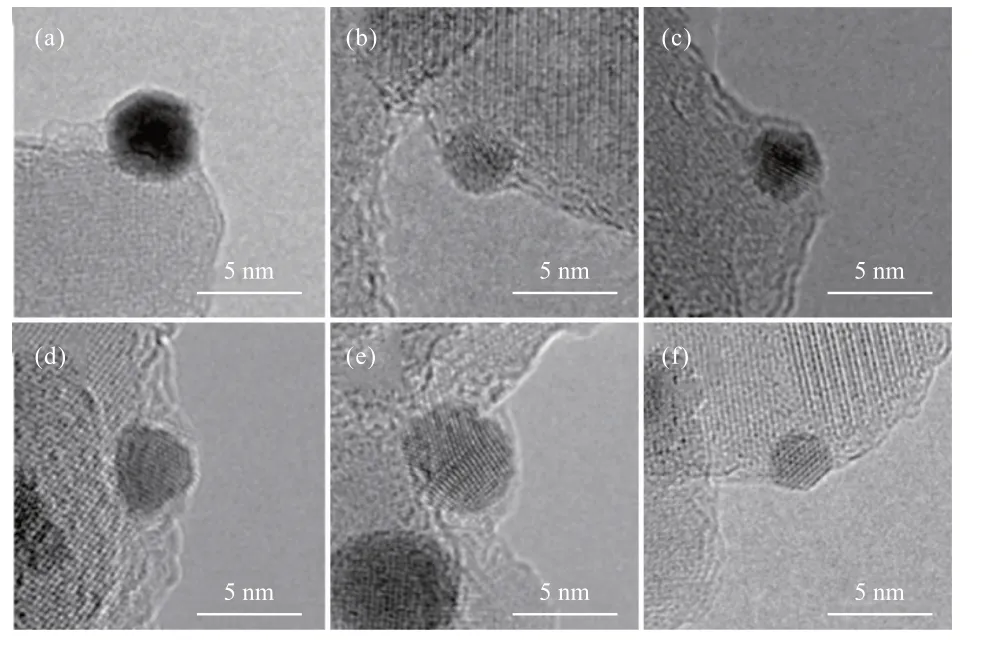
圖2 Au/H-200 (a)、Au/H-300 (b)、Au/H-400 (c)、Au/H-500 (d)、Au/H-600 (e)和Au/H-500-H2 (f)的HRTEM照片[19]Fig.2 HRTEM images of Au/H-200 (a), Au/H-300 (b), Au/H-400 (c),Au/H-500 (d), Au/H-600 (e) and Au/H-500-H2 (f)[19]
除HRTEM之外,其他高分辨電子顯微鏡[21-22],如高角環(huán)形暗場(chǎng)像-掃描透射電子顯微鏡(HAADFSTEM)也可用于觀測(cè)載體與金屬納米顆粒之間的包覆情況。同時(shí),通過(guò)漫反射紅外光譜(DRⅠFT)探測(cè)催化劑對(duì)探針?lè)肿樱ㄈ鏑O、H2)的吸附行為,亦可以側(cè)面說(shuō)明金屬與載體之間的包覆現(xiàn)象,而原位DRⅠFT更是常用于探測(cè)這種包覆的變化,進(jìn)而說(shuō)明相互作用的強(qiáng)弱變化,這部分內(nèi)容將在2.1小節(jié)舉例闡述。
1.2 金屬-載體的化學(xué)鍵表征
很多時(shí)候,金屬-載體之間的相互作用可以用化學(xué)鍵來(lái)描述,特別是對(duì)于SMSⅠ[23]。事實(shí)上,形成化學(xué)鍵是電荷轉(zhuǎn)移的一種表現(xiàn)結(jié)果。例如當(dāng)一個(gè)電子從金屬轉(zhuǎn)移至載體(如金屬氧化物)上時(shí),二者之間就可能形成金屬—氧鍵;而如果金屬-載體之間的相互作用較弱,則可能只有部分電荷偏移。上述兩種情況均普遍存在于MSⅠ的體系中。
X射線吸收精細(xì)結(jié)構(gòu)(XAFS)譜圖包括X射線吸收近邊結(jié)構(gòu)(XANES)譜圖和擴(kuò)展X射線吸收精細(xì)結(jié)構(gòu)(EXAFS)譜圖兩部分,其具有元素分辨性,可以提供凝聚態(tài)物質(zhì)的結(jié)構(gòu)信息[24-25]。通過(guò)XANES譜圖可以獲得材料的對(duì)稱性和化學(xué)態(tài)信息;通過(guò)EXAFS譜圖可以獲得吸收原子的配位環(huán)境參數(shù),如原子種類、配位數(shù)和原子間距等。因此,XAFS經(jīng)常被用于表征金屬-載體的界面相互作用,確定金屬與氧化物載體(如CeO2)之間的鍵合特征[26-28]。例如,2019年,KOTTWⅠTZ等[28]研究了單原子分散的Pt與CeO2載體之間的相互作用,通過(guò)XAFS譜圖結(jié)合計(jì)算模擬,證實(shí)了Pt—O—Ce鍵的形成。CeO2負(fù)載單原子Pt材料(Pt SAC/CeO2)的XANES譜圖與在k空間和R空間的EXAFS譜圖見圖3,同時(shí)為了對(duì)比,圖3提供了Pt金屬箔、α-PtO2和負(fù)載型Pt納米顆粒(Pt NP/CeO2)的數(shù)據(jù)[28]。由圖3可知,納米Pt材料與塊體Pt材料的EXAFS譜圖的特征峰完全不同。進(jìn)一步分析數(shù)據(jù)并進(jìn)行擬合可知,在Pt SAC/CeO2中,與Pt緊鄰的是4個(gè)O,Pt與O之間的距離為1.995 × 10-10m,同時(shí),Ce與Pt的距離為3.340 × 10-10m,這分別與密度泛函理論(DFT)優(yōu)化后的PtCe40O80納米結(jié)構(gòu)模型中Pt—O鍵長(zhǎng)和Pt與Ce的間距接近,證明Pt SAC/CeO2中形成了Pt—O—Ce鍵。

圖3 不同Pt材料的XANES譜圖(a)、k空間(b)和R空間(c)的EXAFS譜圖及Pt SAC/CeO2的實(shí)驗(yàn)與擬合結(jié)果(d)[28]Fig.3 XANES spectra (a), EXAFS spectra in k space (b) and R space (c) of different Pt materials and experimental and fitting results of Pt SAC/CeO2 (d)[28]
此外,振動(dòng)和轉(zhuǎn)動(dòng)光譜用于表征化學(xué)鍵更加直觀。從原理上講,F(xiàn)TⅠR和Raman均可用于表征金屬-載體之間的成鍵情況,如金屬—氧鍵、金屬—金屬鍵等[23,29],二者原理不同但信息相似,在使用過(guò)程中存在一定的互補(bǔ)性。與FTⅠR相比,Raman可以提供低波數(shù)(低于400 cm-1)的振動(dòng)信息、更適用于無(wú)機(jī)材料的檢測(cè),而FTⅠR的探測(cè)范圍通常是400~4000 cm-1,非對(duì)稱分子的紅外信號(hào)比Raman信號(hào)更強(qiáng)。LEE等[29]通過(guò)Raman分析證明,當(dāng)Pt浸漬于CeO2載體之后,Pt通過(guò)Pt—O—Ce鍵錨定在了載體表面,即產(chǎn)生了SMSⅠ。由圖4可知,隨著Pt含量的增加,屬于Pt—O—Ce鍵的新振動(dòng)峰,即567 cm-1(Pt—O—Ce鍵)和655 cm-1(Pt—O鍵)處振動(dòng)峰的振動(dòng)強(qiáng)度逐漸增大。BUKHARⅠ等[30]使用FTⅠR研究了纖維型SBA-15負(fù)載的Ni催化劑的MSⅠ,發(fā)現(xiàn)載體SBA-15中Si—OH鍵振動(dòng)強(qiáng)度的減弱側(cè)面證明了Si—O—Ni鍵的形成。
除此之外,在需要做好急流動(dòng)性參數(shù)控制,在正式操作過(guò)程中,壓漿泵需要始終保持開啟狀態(tài),并在壓漿泵中注入含量適中的水分,不斷攪拌,促使?jié){液最終到達(dá)最佳濃度和黏稠狀態(tài)。當(dāng)漿液黏稠度達(dá)到合適狀態(tài)后,需要用亞水泵將漿液直接輸送到孔洞中進(jìn)行施工。施工過(guò)程需要不斷保證持續(xù)性,施工順序需要按照從上至下一次完成,除此以外,還需要關(guān)注漿體時(shí)間。漿液關(guān)閉時(shí)需要同時(shí)關(guān)閉壓漿泵,如果沒有關(guān)閉,則需要提高0.5MPa壓力,確保壓漿泵可以在自然條件下自動(dòng)停止工作。
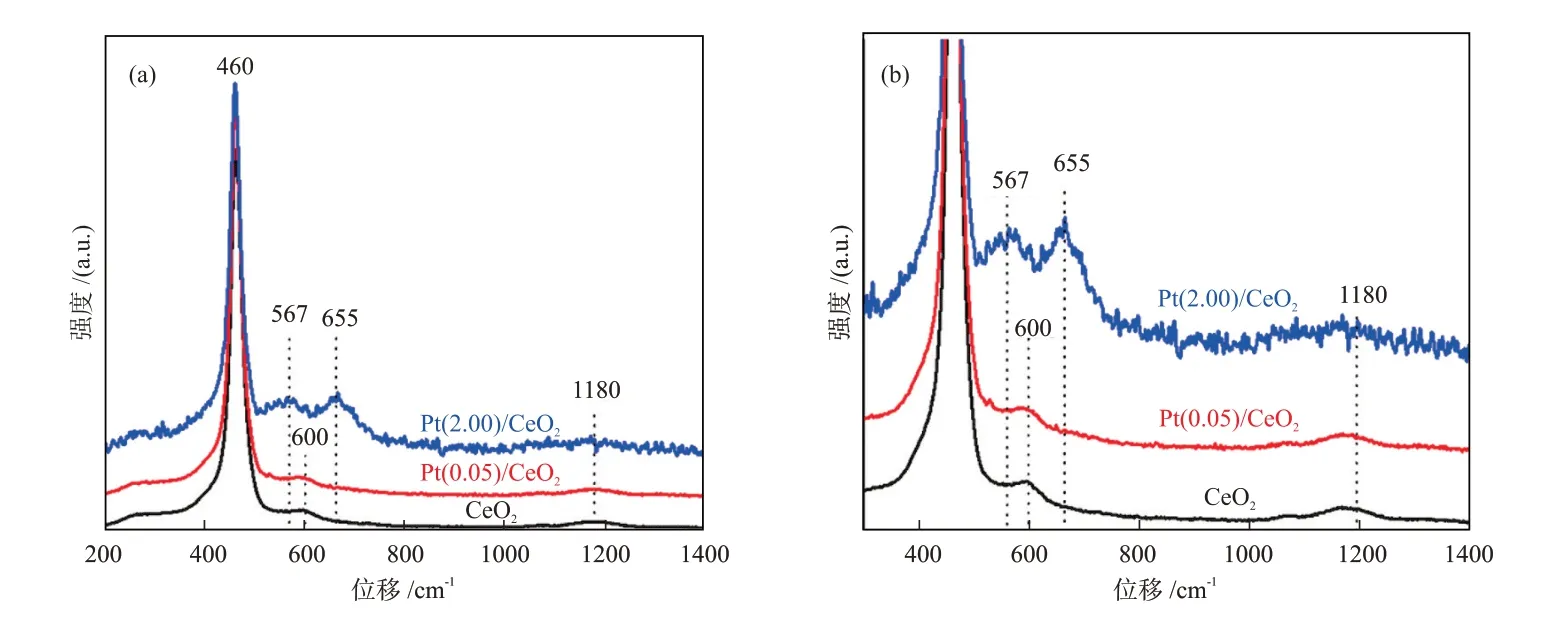
圖4 CeO2、Pt(0.05)/CeO2和Pt(2.00)/CeO2的Raman譜圖(a)和300~1400 cm-1局部放大譜圖(b)[29]。Fig.4 Raman spectra (a) and locally amplified spectra in 300~1400 cm-1 (b) of CeO2, Pt(0.05)/CeO2 and Pt(2.00)/CeO2[29]
1.3 金屬-載體的電荷轉(zhuǎn)移表征
一般地,MSⅠ下的電荷轉(zhuǎn)移表現(xiàn)為表面元素價(jià)態(tài)和電子結(jié)構(gòu)的變化,因此可以使用元素敏感的表征技術(shù)進(jìn)行檢測(cè),如XPS、紫外光電子能譜(UPS)、俄歇譜(AES)等[31-35]。例如,LU等[36]研究了一系列貴金屬納米催化劑(Pt、Pd、Rh、Ⅰr和Ag)與TiO2載體之間的相互作用,部分研究結(jié)果見圖5。由圖5可知,對(duì)于Pt/TiO2,電子結(jié)合能位于71.0 eV、71.9 eV和72.9 eV處的XPS峰分別歸屬于Pt0、Pt2+和Pt4+,而Pt0的XPS峰一般出現(xiàn)在71.5 eV,即在該催化劑中,Pt0的XPS峰向低能量偏移,說(shuō)明Pt帶負(fù)電荷。Pd/TiO2也表現(xiàn)出類似的XPS峰偏移,表明Pt/TiO2和Pd/TiO2通過(guò)MSⅠ誘導(dǎo)TiO2局部電荷轉(zhuǎn)移到貴金屬納米顆粒上,這種電荷轉(zhuǎn)移使Pt/TiO2和Pd/TiO2比另幾種貴金屬催化劑表現(xiàn)出更好的催化性能。

圖5 M/TiO2的XPS譜圖[36]Fig.5 XPS spectra of M/TiO2[36]
此外,電荷轉(zhuǎn)移必然會(huì)改變載體的局部配位環(huán)境,氧化物載體的電子結(jié)構(gòu)擾動(dòng)可通過(guò)EPR進(jìn)行表征[37-39]。WANG等[40]采集了ZnO和Ni/ZnO樣品的EPR譜圖并探究了Ni和ZnO之間電荷轉(zhuǎn)移路徑以及SMSⅠ作用對(duì)CO甲烷化反應(yīng)的影響(圖6)。ZnO晶格中存在VOo、VO˙和VO˙ 3種氧空位,其中只有VO˙含未成對(duì)電子,具有EPR活性。由圖6可知,隨著還原溫度的升高,與ZnO的EPR信號(hào)(歸屬于VO˙)相比,Ni/ZnO的信號(hào)逐漸減弱,說(shuō)明ZnO載體的VO˙失去電子或得到電子,形成了沒有EPR活性的VOo或VO˙。通過(guò)XPS譜進(jìn)一步確認(rèn)可知,Ni和ZnO粒子之間最可能的電子轉(zhuǎn)移路徑是ZnO晶格中的VO˙將電子傳遞給Ni,形成VOo。
用DRⅠFT表征催化劑對(duì)探針?lè)肿樱ㄈ鏑O)的吸附行為,也可反映催化劑表面的電荷分布情況,例如,CO—Au鍵的紅外吸收帶從2101 cm-1藍(lán)移至2113 cm-1,表明Au納米顆粒表面正電荷增加[20]。
以上主要從MSⅠ的靜態(tài)性質(zhì)展開,分別介紹了金屬-載體界面微觀結(jié)構(gòu)、二者之間成鍵和電荷轉(zhuǎn)移情況的常用非原位表征方法,其總結(jié)見表1。

表1 MSI的非原位表征方法總結(jié)Table 1 Summary of ex situ characterization methods for MSI
2 MSⅠ的原位表征
MSⅠ具有可逆性,催化劑預(yù)處理?xiàng)l件和反應(yīng)條件均可能使相互作用發(fā)生變化。因此,在預(yù)處理過(guò)程和反應(yīng)過(guò)程中對(duì)MSⅠ進(jìn)行原位動(dòng)態(tài)表征十分必要。一方面有利于理解相互作用的產(chǎn)生機(jī)制,另一方面有利于深入探究相互作用對(duì)于催化活性和催化反應(yīng)機(jī)制的影響。基于原位表征技術(shù),人們對(duì)MSⅠ的強(qiáng)弱變化、反應(yīng)過(guò)程中的界面結(jié)構(gòu)和組分變化,以及電荷轉(zhuǎn)移的動(dòng)態(tài)過(guò)程展開了研究。
2.1 MSI的強(qiáng)弱變化表征
催化劑的MSⅠ增強(qiáng)會(huì)導(dǎo)致其對(duì)小分子的吸附減弱。因此當(dāng)選擇合適的探針?lè)肿訒r(shí),原位DRⅠFTS可以敏銳地探測(cè)出金屬表面的吸附性能,是表征MSⅠ強(qiáng)弱變化最常用的手段[31,41-43]。TANG等[44]分別研究了Au納米顆粒與TiO2(Au/T)、HAP(Au/H)和TiO2/HAP(Au/TH)載體之間的相互作用情況。由圖7(a)可知,相比于未煅燒的樣品(Au/載體-uc),800 ℃煅燒之后的3種Au催化劑(Au/載體-800)的CO振動(dòng)峰均大幅減弱,說(shuō)明此時(shí)載體對(duì)Au納米顆粒形成包覆,Au與載體TiO2和HAP之間均存在強(qiáng)相互作用。進(jìn)一步地,在溫度為-130 ℃時(shí),對(duì)預(yù)吸附了CO的Au/TH-800樣品通入O2,采集了30 min內(nèi)的原位DRⅠFT譜圖(圖7(b))。由圖7(b)可知,吸附于TiO2上的CO峰(CO/TiO2)的強(qiáng)度減弱,CO/Au峰的強(qiáng)度不變,CO2振動(dòng)峰的強(qiáng)度增大,說(shuō)明載體中引入的TiO2有利于CO催化氧化反應(yīng)。
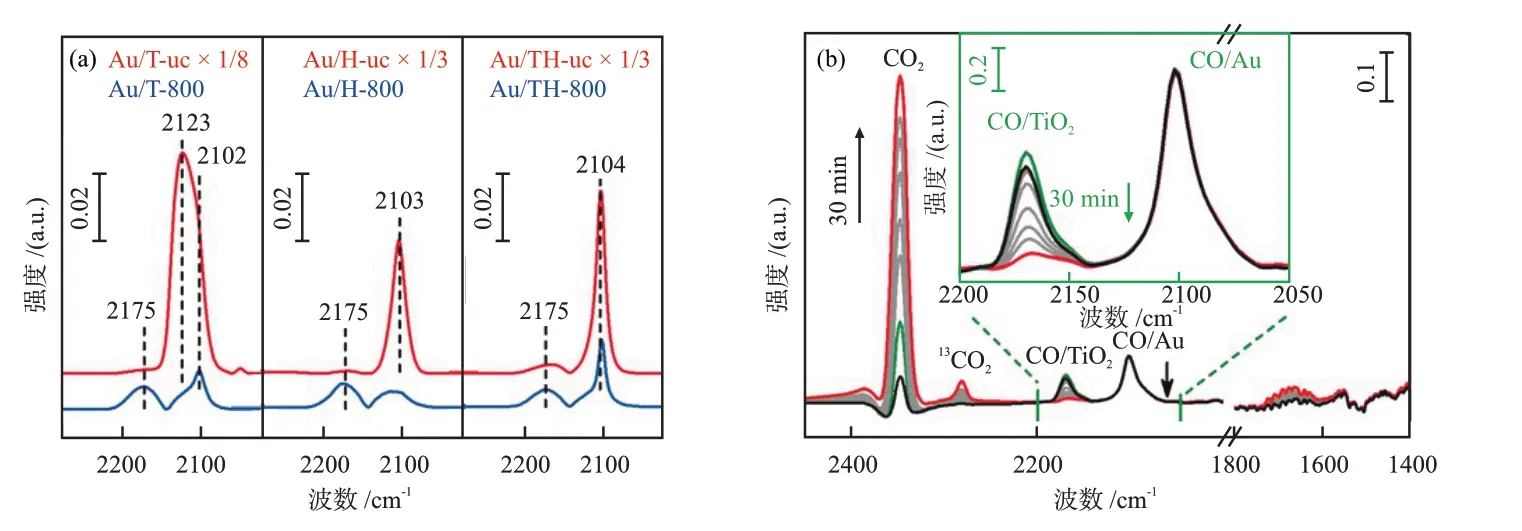
圖7 吸附CO 10 min后不同Au催化劑的原位DRIFT譜圖(室溫)(a)和在預(yù)吸附了CO的Au/TH-800中通入2.0 kPa O2后Au/TH-800的原位DRIFT譜圖(-130 ℃)(b)[44]Fig.7 In situ DRIFT spectra of different Au catalysts after 10 min of CO adsorption (room temperature) (a) and in situ DRIFT spectra of Au/TH-800 pre adsorbed with CO after introducing 2.0 kPa O2 (-130 ℃) (b)[44]
此外,H2程序升溫還原(H2-TPR)也是一種常見的表征相互作用強(qiáng)弱的手段。例如,銅基催化劑的H2還原溫度較純CuO相比有一定幅度的下降,由此可說(shuō)明存在MSⅠ。CAO等[45]通過(guò)H2-TPR研究了負(fù)載CeO2、ZrO2、CeO2-ZrO2的Cu催化劑,發(fā)現(xiàn)CuCeZr、CuCe和CuZr催化劑的H2還原峰分別位于142 °C、154 °C和162 °C,表明3種催化劑中Cu與載體的相互作用依次減弱。當(dāng)然,H2-TPR是一種非原位方法,這里僅作為補(bǔ)充方法進(jìn)行說(shuō)明,H2-TPR只能表征催化劑的相互作用強(qiáng)弱,而不像原位DRⅠFTS可以表征強(qiáng)弱變化,但H2-TPR是催化劑的常用表征手段,比原位DRⅠFTS更為經(jīng)濟(jì)。
2.2 金屬-載體之間的界面結(jié)構(gòu)變化表征
基于原位電鏡、原位X射線譜學(xué)等手段,可以實(shí)時(shí)觀測(cè)催化劑的結(jié)構(gòu)演變[46-52]。DONG等[49]發(fā)現(xiàn)在甲烷干重整(DRM)反應(yīng)中,弱氧化性氣體可誘導(dǎo)Ni納米顆粒與六方氮化硼(h-BN)納米片之間的相互作用,這種相互作用來(lái)源于氣體刻蝕h-BN載體產(chǎn)生的超薄氧化硼(BOx)覆蓋層對(duì)Ni顆粒的包覆。原位環(huán)境掃描透射電鏡(ESTEM)展現(xiàn)了CO2(約3.5 Pa)對(duì)h-BN的原位刻蝕過(guò)程,通過(guò)原位場(chǎng)發(fā)射掃描電子顯微鏡(FESEM)對(duì)比了800 ℃時(shí)不同氣體(H2、CO2、H2O和O2)對(duì)MSⅠ的影響,發(fā)現(xiàn)弱氧化性氣體(CO2、H2O)可以促進(jìn)Ni與h-BN的界面反應(yīng)(圖8)[49]。

圖8 100 ℃ (a)和676 ℃ (b)下Ni/h-BN在CO2中的原位ESTEM照片及800 ℃下Ni/h-BN在H2、CO2、H2O 和O2中的原位FESEM照片(c)[49]Fig.8 In situ ESTEM images of Ni/h-BN in CO2 at 100 ℃ (a)and 676 ℃ (b) and in situ ESTEM images of Ni/h-BN in H2, CO2, H2O and O2 at 800 ℃ (c)[49]
BECK等[48]基于原位電鏡、原位XPS和原位XRD,輔以DFT計(jì)算,研究了Pt與TiO2載體相互作用的形成過(guò)程和結(jié)構(gòu)變化機(jī)理,揭示了H2和O2在該過(guò)程中的作用。先在600 ℃、100 kPa H2氛圍中對(duì)樣品進(jìn)行還原,然后在O2氛圍下處理,之后再次轉(zhuǎn)換為H2氛圍。對(duì)上述處理過(guò)程進(jìn)行原位表征可知,H2氛圍下,TiO2遷移形成覆蓋層和形成Pt-Ti合金是互相競(jìng)爭(zhēng)的過(guò)程,將樣品暴露于O2氛圍中之后,Ti會(huì)從合金中分離出來(lái),形成更厚的TiO2覆蓋層。這種厚覆蓋層在O2中很穩(wěn)定,表明MSⅠ也可應(yīng)用于催化氧化反應(yīng)。
2.3 金屬-載體之間的電荷轉(zhuǎn)移過(guò)程表征
類似于靜態(tài)電荷轉(zhuǎn)移的表征思路,基于原位XAFS、氣氛條件下的XPS(AP-XPS)等技術(shù),可以考察反應(yīng)條件下由MSⅠ引起的催化劑表面電性變化[47,53-60]。LⅠU等[47]探究了一種CeO2負(fù)載的Ru納米團(tuán)簇用于DRM反應(yīng)的催化活性和穩(wěn)定性,發(fā)現(xiàn)MSⅠ改變了Ru納米團(tuán)簇的電子性質(zhì),電子轉(zhuǎn)移形成的Ru*-CeO2-x保證了催化活性。在CH4還原氣氛中,由原位Ru K吸收邊XANES譜圖(圖9(a))可知,隨著溫度的提高,RuO2逐漸被還原為Ru,并在150~200 ℃發(fā)生了快速還原過(guò)程(圖9(b))。類似的還原過(guò)程也出現(xiàn)在CeO2載體表面,由原位AP-XPS譜圖(圖9(c))可知,從150 ℃開始,載體表面逐漸產(chǎn)生Ce3+,同時(shí)還原過(guò)程伴隨著氧化物載體中晶格氧的減少。
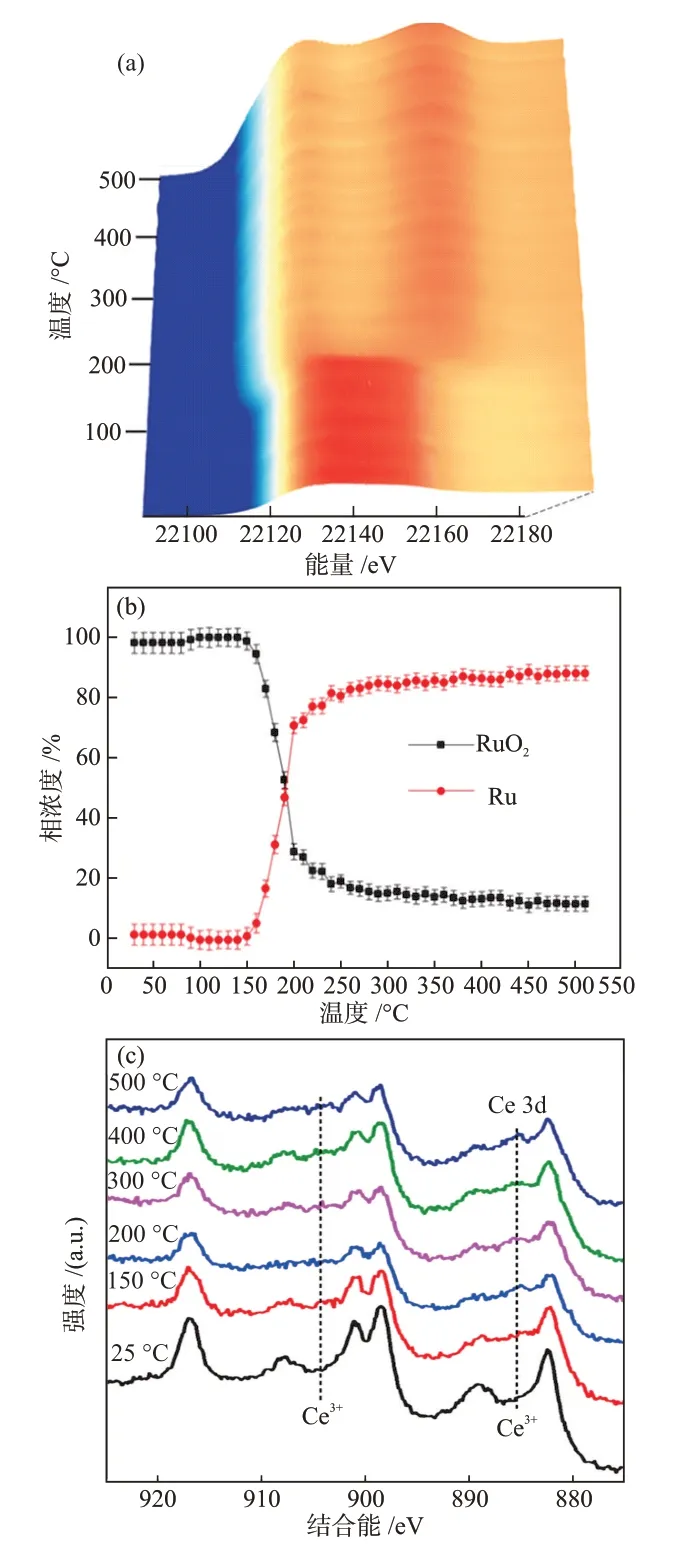
圖9 Ru(NC)/CeO2與CH4反應(yīng)時(shí)的原位Ru K吸收邊XANES譜圖(a)和對(duì)應(yīng)的擬合結(jié)果(b)及6.67 Pa CH4作用下Ru(NC)/CeO2的原位AP-XPS譜圖(c)[47]Fig.9 In situ Ru K-edge XANES spectra during reaction of Ru(NC)/CeO2 with CH4 and corresponding fitting results (b) and in situ AP-XPS spectra of Ru(NC)/CeO2 under 6.67 Pa of CH4[47]
同樣,通過(guò)原位EPR亦能夠監(jiān)測(cè)催化反應(yīng)過(guò)程中金屬離子的價(jià)態(tài)變化,進(jìn)而得到電荷轉(zhuǎn)移的過(guò)程信息。如WANG等[55]基于原位EPR輔以一系列非原位表征手段,研究了富氫中CO優(yōu)先氧化(PROX)反應(yīng)過(guò)程中的CuO-CeO2催化劑的氧化還原行為,認(rèn)為協(xié)同氧化還原是主要的反應(yīng)機(jī)制,即Ce4+被還原為Ce3+伴隨著Cu0和Cu+被氧化為Cu2+,這一機(jī)制表明了Cu與Ce之間的相互作用對(duì)催化性能有重要影響。
需要注意,上述原位表征方法更側(cè)重的是不同反應(yīng)條件下或?qū)嶋H反應(yīng)過(guò)程中金屬-載體之間的電荷轉(zhuǎn)移過(guò)程,實(shí)際上探測(cè)的是電荷轉(zhuǎn)移之后的結(jié)果狀態(tài)(如金屬價(jià)態(tài))。而真正意義上的電荷轉(zhuǎn)移過(guò)程是非常迅速的,其時(shí)間尺度一般在飛秒、皮秒量級(jí),這種時(shí)間尺度的表征則需要借助時(shí)間分辨光譜技術(shù)。
本節(jié)主要介紹了催化劑預(yù)處理?xiàng)l件和反應(yīng)條件下,MSⅠ可能產(chǎn)生的動(dòng)態(tài)變化,以及表征這類動(dòng)態(tài)性質(zhì)的常用原位技術(shù),各原位表征方法的特征總結(jié)見表2。通過(guò)原位表征技術(shù),可以獲取特定條件下MSⅠ的強(qiáng)弱和界面微觀結(jié)構(gòu)的變化、電荷轉(zhuǎn)移的動(dòng)態(tài)過(guò)程等信息,進(jìn)而理解相互作用和催化反應(yīng)的機(jī)理。目前原位表征的技術(shù)和經(jīng)濟(jì)門檻均比較高,且大部分原位技術(shù)仍然只能滿足特定條件下的探測(cè),較難實(shí)現(xiàn)任意或?qū)嶋H反應(yīng)條件下的實(shí)時(shí)觀測(cè),但是隨著技術(shù)的飛速迭代和發(fā)展,原位表征方法將不斷成熟和普及。

表2 MSI的原位表征方法總結(jié)Table 2 Summary of in situ characterization methods for MSI
3 結(jié)語(yǔ)與展望
MSⅠ的宏觀概念并不復(fù)雜,但是微觀細(xì)節(jié)比較豐富,隨著研究者對(duì)體系認(rèn)識(shí)的逐漸深入,目前已發(fā)展出豐富多樣的配套表征方法和策略。事實(shí)上,任何一種表征手段都有其特定的使用場(chǎng)景和優(yōu)缺點(diǎn),本文根據(jù)MSⅠ的不同表現(xiàn)特征對(duì)表征技術(shù)進(jìn)行了簡(jiǎn)單分組,同一種技術(shù)經(jīng)常可以給出不同方面的信息,且MSⅠ的各個(gè)特征之間本身也存在內(nèi)在聯(lián)系。因此,多種表征手段結(jié)合的方式才能實(shí)現(xiàn)對(duì)體系較全面的認(rèn)識(shí)。
目前,非原位表征方法已經(jīng)相當(dāng)成熟,相比原位表征,其使用成本和技術(shù)門檻均較低,筆者認(rèn)為,關(guān)于MSⅠ的非原位表征,未來(lái)可發(fā)展的部分在于更高的空間分辨率,即對(duì)微小區(qū)域內(nèi)的組分間相互作用進(jìn)行表征,如金屬原子和化學(xué)鍵的可視化。但是,非原位表征技術(shù)只能給出MSⅠ的靜態(tài)信息,例如預(yù)處理或催化反應(yīng)前后催化劑的形貌、活性金屬與載體的價(jià)態(tài)及電荷分布情況等,而無(wú)法表征實(shí)際反應(yīng)過(guò)程中相互作用的動(dòng)態(tài)變化。理論上,原位表征技術(shù)可以彌補(bǔ)這一缺陷,給出特定條件下MSⅠ強(qiáng)弱的實(shí)時(shí)變化、電荷轉(zhuǎn)移的動(dòng)態(tài)過(guò)程等信息,進(jìn)而有助于探究MSⅠ的機(jī)理和催化反應(yīng)機(jī)理。原位動(dòng)態(tài)表征技術(shù)的發(fā)展及其在該領(lǐng)域中的應(yīng)用,還有很長(zhǎng)的路要走。因受限于儀器使用條件,如特定壓力和溫度范圍、特定反應(yīng)氣氛和特定反應(yīng)狀態(tài)等,目前大部分原位表征技術(shù)只能滿足特定催化反應(yīng)的動(dòng)態(tài)表征,因此,如何在實(shí)際反應(yīng)條件下進(jìn)行原位表征是未來(lái)技術(shù)需要解決的問(wèn)題。同時(shí),提高分析方法的時(shí)間分辨率,實(shí)現(xiàn)真正意義上的動(dòng)態(tài)表征,也是未來(lái)研究者們努力的方向。
猜你喜歡
當(dāng)代陜西(2020年13期)2020-08-24 08:22:02
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
制造技術(shù)與機(jī)床(2017年5期)2018-01-19 02:49:17
金秋(2017年4期)2017-06-07 08:22:16
中國(guó)材料進(jìn)展(2016年10期)2016-12-26 06:50:20
濰坊學(xué)院學(xué)報(bào)(2016年2期)2016-12-01 13:00:11
浙江大學(xué)學(xué)報(bào)(工學(xué)版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
中國(guó)資源綜合利用(2016年4期)2016-01-22 08:27:23
