污水深度處理中常見固相臭氧催化劑及其催化機理綜述
2024-03-22 10:15:58李旭芳沈鵬飛馬魯銘
凈水技術 2024年3期
李旭芳,沈鵬飛,*,馬魯銘
(1.中國鐵工投資建設集團有限公司,北京 101300;2.中國中鐵生態環境專業研發中心,上海 200331;3.同濟大學環境科學與工程學院,上海 200092)
在臭氧(O3)氧化工藝中,催化劑的使用可促進反應過程中·OH的產生,從而克服單獨O3氧化的選擇性,提高污水深度處理的效果。固相催化劑具有固液分離效果好、二次污染少、可循環使用等優點,在實際應用中有較好的適用性,因此得到學界和工業界的重點關注。固相催化劑的催化效果很大程度上取決于催化劑的種類、催化劑表面活性位點及液相組成與催化劑之間的相互作用。因此,不同催化劑的催化機理存在較大差異,需要分別討論。
主要的固相催化劑包括金屬及金屬氧化物、金屬及其氧化物的負載和碳基材料及其負載型催化劑[1]。本文僅考慮催化劑有效成分的催化機理,有關金屬氧化物負載于其他材料或金屬氧化物作為載體負載其他有效成分的催化劑,暫不在本文的重點討論范圍內。因此,本文著重于總結分析金屬及其氧化物、碳基材料的催化O3氧化效果及其催化機理。根據目前理論研究進展、材料的易得性及可預見的應用前景,本文重點關注的金屬及其氧化物是鐵(Fe)、錳(Mn)、鈰(Ce)、鎳(Ni)、鈷(Co)、鈦(Ti)及其氧化物,碳基材料是活性炭、碳納米管和石墨烯。
1 金屬氧化物
1.1 鐵氧化物
Fe及其氧化物在自然界中豐度較大、易于合成、活性較強且幾乎無毒無害,在水處理中應用廣泛。Fe及Fe的氧化物包括零價鐵(Fe0)、Fe2O3、Fe3O4、FeOOH等,因其具有活潑的化學特性及豐富的反應活性位點,均可以表現出較好的O3催化效果[2]。研究[3]表明,Fe0與O3的耦合工藝能夠獲得協同效果,在廢水處理過程中能夠取得更高的TOC、CODCr去除率,同時提高廢水的可生化性,同步去除渾濁度、色度等;Wu等[4]將鐵刨花應用于印染廢水的深度處理,發現在催化O3氧化過程結束后,100%的蛋白質和42%的多糖被降解,廢水可生化性大幅提高、生物毒性降低,表明該工藝既能夠作為廢水處理的最終步驟以滿足CODCr的排放需求,又能夠為進一步生化處理提供有利條件。
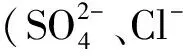
相比之下,Fe2O3和Fe3O4的催化效果較弱。Trapido等[11]對比研究了多種過渡金屬氧化物催化O3氧化間二硝基苯的效果,發現Fe2O3能夠顯著提高間二硝基苯的降解速率,但提高CODCr去除率的能力較其他金屬氧化物(Ni2O3等)不明顯。Zhu等[2]合成介孔Fe3O4用于催化O3氧化阿特拉津,反應10 min后阿特拉津去除率由單獨O3的9%提高至82%;但TOC去除率較低,不足20%。因此,Fe2O3和Fe3O4僅能加速某類物質的降解,而不能實現有機物礦化。
Fe的不同價態及其組成會形成不同的晶體結構,物理化學性質差異較大。如FeO非常不穩定,極易被氧化為Fe2O3或Fe3O4;Fe3O4有極強的磁性,從而提供了額外的回收和固液分離方法;Fe2O3表面有豐富的酸性位點;FeOOH的表面羥基基團密度更大。同時,Fe0是高效還原劑,極易被氧化為Fe(Ⅱ)、Fe(Ⅲ)、Fe(Ⅳ)等多種價態,在價態變化的過程中可實現高效的電子轉移,從而可能成為促進O3分解的催化劑。Fe及其氧化物由于自身性質多樣、目標污染物差異及試驗條件的改變而有不同的催化機制,目前尚未有統一的定論,但各種論點均有兩個共同點:一是提高O3傳質效率,二是促進·OH的產生。催化過程主要涉及4個方面[12]。
其一是Fe(Ⅱ)/Fe(Ⅲ)的均相催化作用,如式(1)~式(6)。
(1)
(2)
Fe3++O3+H2O→FeO2++·OH+O2+H+
(3)

(4)
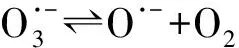
(5)
O·-+H2O·OH+OH-
(6)

其三可能是芬頓反應的貢獻,如式(7)和式(8)[15]。
Fe+O2+2H+→Fe2++H2O2
(7)
Fe2++H2O2→Fe3++·OH+OH-
(8)
其四是反應過程中的混凝沉淀作用。鐵氧化物溶出的Fe2+/Fe3+在水環境中生成Fe(OH)2或Fe(OH)3絮體,可作為混凝劑,通過混凝沉淀作用去除有機物。
1.2 錳氧化物
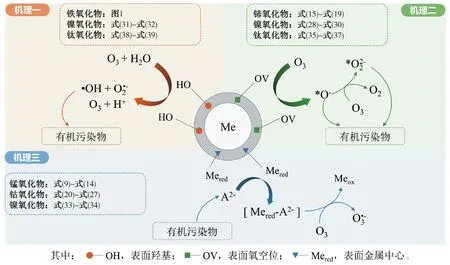
作為O3催化劑的錳氧化物主要包括MnO2和Mn3O4。MnO2是氣相O3分解最有效的固相催化劑,因此,錳氧化物在水環境中催化O3的效果也引起了廣泛關注。但是不同的研究,結果差異較大。Faria等[16]將合成Mn3O4作為催化劑,其催化O3氧化苯胺和磺胺酸的效果都很顯著,TOC去除率相對單獨O3分別增加了35%和10%,兩者的最終降解產物中均有草酸等小分子酸。Ma等[17]證明,只需極小劑量的Mn(Ⅱ)即可與O3原位生成MnO2以發揮催化作用,顯著提高阿特拉津去除率;通過高錳酸氧化Mn(Ⅱ)預先合成的MnO2亦有相似的催化效果;但商用MnO2沒有任何催化作用。Trapido等[11]考察了多種過渡金屬離子及其氧化物對間二硝基苯的催化O3氧化效果,結果表明,在O3進氣質量濃度為12 mg/L時,各種氧化物催化效果排序如下:單獨O3≈MnO2≈Cr2O3 在MnO2催化O3過程中,pH和有機物種類是主要的影響因素。Tong等[18]研究了β-MnO2和γ-MnO2的催化效果,發現pH值= 1時磺基水楊酸的TOC去除率相對單獨O3均提高了25%,而在pH值=6.8時幾乎無催化作用;磺基水楊酸被O3直接氧化可生成草酸,催化O3可有效降解草酸,而單獨O3氧化時草酸濃度不斷積累;但當有機物為丙酸時,MnO2在任何pH條件下均無催化作用。此外,MnO2的催化作用與其結構性質有一定關系,Nawaz等[19]利用介孔α-MnO2催化O3氧化4-氯酚,TOC去除率相比商用α-MnO2提高了15%;Dong[20]等合成的β-MnO2有較大的比表面積和結構優勢,在不吸附有機物的條件下亦可大幅提高苯酚的CODCr去除率。 (9) (10) Mn(Ⅱ)+Mn(Ⅳ)→2Mn(Ⅲ) (11) Mn(Ⅱ)+O3+2H+→Mn(Ⅳ)+O2+H2O (12) 在弱酸性條件下(pH值為4~6),草酸與Mn(Ⅲ)形成雙分子或三分子復合物,但該復合物的反應活性遠低于單分子復合物;MnO2的加入僅可活化草酸分子,生成更易被氧化的AO-[式(13)];此時O3參與草酸的氧化,且可能生成·OH[式(14)]。 (13) (14) 綜上,MnO2在水環境中催化O3氧化有機物時,有機物的降解或自由基的產生均以形成表面復合物為前提。以上機理可較完整地解釋MnO2催化O3氧化對有機物的選擇性,即是否能與Mn(Ⅲ)生成表面復合物。 作為O3催化劑的鈰氧化物主要是CeO2。CeO2是立方螢石結構氧化物,即一個鈰原子周圍環繞8個氧原子,是最重要的稀土元素氧化物。CeO2中的鈰原子有多個價態[Ce(Ⅲ)/Ce(Ⅳ)],在Ce被還原時可形成氧空位來存儲和釋放氧,常用來作為催化劑、助催化劑及催化劑載體。研究表明,鈰氧化物在O3氧化過程中具有較好的催化性能。Orge等[22]研究了CeO2及鈰基混合氧化物催化O3氧化羧酸及芳香環類污染物(草酸、苯胺及染料等)的效果,TOC去除率均提高了至少50%,表明CeO2催化O3氧化對有機物種類無選擇性。Mena等[23]發現CeO2的催化效果與其形貌及結構特征有較大關系,納米棒狀材料具有更大的比表面積,導致具有更多的表面晶格缺陷和氧空位,催化O3效果更好。Esmalipour等[24]用紫外光預處理CeO2以促進表面氧空位的生成,發現預處理之后的催化劑對水楊酸的催化O3氧化效果提高了3倍,再次證實氧空位的重要性。除此之外,污染物去除效果還與表面Ce(Ⅲ)的百分比有關。一般來講,Ce(Ⅲ)比Ce(Ⅳ)更穩定,但在CeO2中Ce(Ⅳ)占主導位置;催化反應發生時,Ce(Ⅲ)首先被氧化為較不穩定的Ce(Ⅳ),再被有機物或其他部分的電子還原為Ce(Ⅲ)[25]。Orge等[26]用不同方法合成納米結構CeO2,發現隨著材料表面Ce(Ⅲ)百分比增加,其催化O3礦化草酸的程度越高,催化效果越好。 表面氧空位和氧化還原電對[Ce(Ⅲ)/Ce(Ⅳ)]是CeO2催化O3的主要活性位點[29]。CeO2催化O3的過程可能主要包括4個步驟[24]:一是O3分子吸附于表面氧空位并分解為表面原子氧[式(15)],可氧化吸附于催化劑表面的污染物,同時可被O3繼續氧化為表面過氧[式(16)];二是Ce(Ⅲ)作為Lewis酸性位點吸附H2O形成表面羥基,O3通過靜電力/氫鍵與表面羥基基團結合并分解為HO2·、HO3·、·OH等自由基,同時Ce(Ⅲ)被氧化為Ce(Ⅳ)[式(17)];三是為了維持表面電荷平衡,Ce(Ⅳ)被表面晶格氧重新還原為Ce(Ⅲ),氧空位重新產生,增加催化劑表面吸附氧的能力[式(18)];最后,O3接受電子被還原為晶格氧,補償催化劑表面的氧缺陷[式(19)]。在這一過程中,Ce(Ⅲ)向O3的電子轉移導致活性氧物種的產生,表面晶格氧參與完成Ce(Ⅲ)/Ce(Ⅳ)的循環,因此,Ce(Ⅲ)/Ce(Ⅳ)和O2-/O2的氧化還原對及表面靜電平衡是提高CeO2催化活性的主要因素。 CeO2+O3→CeO2-O+O2 (15) CeO2-O+O3→CeO2-O2+O2 (16) 2Ce(Ⅲ)-OH+O3→2Ce(Ⅳ)+O2-+2·OH+O2 (17) 4Ce(Ⅳ)+2O2-→4Ce(Ⅲ)+O2 (18) O3+2e-→O2-+O2 (19) 作為O3催化劑的鈷氧化物主要包括CoO、Co3O4等。鈷氧化物是P型半導體,可將O2吸附在高價金屬上生成表面原子氧,在氣相O3分解過程中有顯著催化作用。Dong等[30]合成Co3O4納米顆粒用于催化O3氧化苯酚,其催化效果與顆粒粒徑有關,粒徑越小,比表面積越大、分散性越好、催化效果越好,CODCr去除率由單獨O3的36.2%增加至53.3%。Xu等[31]發現合成β-Co(OH)2可大幅促進O3衰減和對氯硝基苯的降解,但隨著循環使用次數的增加,催化效果明顯降低。鈷氧化物對O3氧化有一定的催化效果,但在加入叔丁醇(TBA)作為·OH抑制劑后,草酸去除率和O3衰減速率均未降低,表明·OH作為主要氧化物種的可能性不大。但是,亦有學者指出鈷氧化物的催化效果不明顯。Gruttadauria等[32]研究了不同結構、形貌和還原特性的鈷催化劑(Co3O4、CoO、CoOx-CeO2),發現其催化效果與表面分散Co(Ⅱ)十分相關,但三者的催化效果都不理想,有較多金屬溶出的狀態下才有一定的催化能力。 (20) (21) (22) (23) Co(Ⅲ)+O3→Co(Ⅲ)-O3 (24) 2Co(Ⅲ)-O3+Co(Ⅱ)(C2O4)→Co3O4+2CO2+O2 (25) Co(Ⅱ)+O3+H2O→Co(Ⅲ)-OH+·OH+O2 (26) (27) NiO+O3→NiO-O3→NiO-O+O2 (28) NiO-O+H2O→NiO+2·OH (29) (30) NiO+H2O→NiO-OH2 (31) NiO-OH2+O3→NiO+HO3·+HO· (32) (33) NiO+-C=C-/-COOH/-C=O→ NiO-C=C-/-COOH/-C=O (34) 作為O3催化劑的鈦氧化物主要是TiO2。TiO2廣泛應用于光催化、光伏電池、氣體傳感器、光敏材料及生物材料等,由于其相對廉價易得且毒性較小,在水溶液中催化O3的應用也得到了廣泛研究。Molnar等[38]將TiO2應用于催化O3氧化飲用水源水中有機質的去除及消毒副產物的產生,表明催化O3條件下TOC去除率可提高約20%,腐植酸類物質可被完全去除。Betrn等[39]系統研究了TiO2存在條件下O3氧化草酸的化學動力學和影響因素,發現表觀反應速率常數可提高約3個數量級。Yang等[40]指出TiO2在中性和酸性條件下可顯著提高阿特拉津的去除率;由于阿特拉津自身具有疏水性,幾乎不會吸附于催化劑表面,證明有機物的氧化作用主要發生在液相主體。Yang等[41]指出TiO2可催化O3氧化硝基苯,同時抑制試驗及EPR[5,5-二甲基-1-吡咯啉-N-氧化物(DMPO)]檢測均證明納米TiO2可促進O3氧化體系中·OH的產生,證實了·OH機理的主導地位。 TiO2催化性能與晶相結構有關。TiO2有4種物質形態:板鈦礦(正交晶系)、銳鈦礦(四方晶系)、金紅石(四方晶系)和TiO2-B(單斜晶系)。Song等[42]研究表明,金紅石比銳鈦礦有更好的催化性能,單位比表面積有機物去除率與TiO2晶體中金紅石的比例成正比。金紅石表面因橋氧原子缺失形成五度配位Ti,比銳鈦礦中四度配位Ti形成的氧空位更穩定;同時,有機物礦化效果與催化劑表面羥基密度正相關,而金紅石表面Ti3+位點更多,氧空位更多,表面羥基密度更大,催化性能更好。TiO2催化性能與晶面表面能相關。就銳鈦礦而言,不同晶面的表面能不同,其催化效果差距很大,(001)晶面的催化效果遠高于(101)晶面,因為H2O分子可在(001)晶面解離為表面羥基,而在(101)晶面沒有反應活性;然而,(101)晶面熱力學穩定性更好,多數銳鈦礦均以(101)晶面為主,催化效果較差[43]。 TiO2催化性能主要源于晶格中的氧空位。銳鈦礦和金紅石晶格中均存在氧空位,O3或H2O分子吸附于氧空位后解離為表面氧原子或·OH,氧化溶液或催化劑表面吸附的有機物[44],其催化機制如下[39,45]。(1)表面氧原子作為氧化物種:首先,O3分子通過一端氧原子與TiO2表面結合,被活化后生成表面氧原子,同時釋放出O2[式(35)];同時,TiO2表面吸附有機物分子[式(36)],被表面氧原子氧化,反應在表面發生[式(37)]。(2)·OH作為氧化物種:在酸性和中性條件下,O3在催化劑表面解離生成·OH[式(38)],反應發生在催化劑表面或液相主體;堿性條件下沒有活性物種生成,TiO2的催化效果較弱[式(39)]。 O3+TiO2?O=O-O-TiO2?O-TiO2+O2 (35) TiO2+[Org]?TiO2-[Org] (36) TiO2-[Org]+O-TiO2?TiO2+CO2+H2O (37) O3+TiO2+H2O→O3-TiO2-H2O→TiO2+O2+2·OH (38) O3+TiO2+OH-→O3-TiO2-OH-→ (39) 從上述分析可知,金屬及其氧化物催化O3的機理大致可分為3類(圖2)。 圖2 金屬及其氧化物催化O3機理 機理二:以表面吸附活性氧原子作為主要氧化劑,如鈰氧化物、鈦氧化物等。 機理三:表面金屬-有機復合產物生成的非自由基機理,如錳氧化物、鈷氧化物等。 對某一金屬氧化物,以上3種機理可單獨作用,亦可同時出現。機理二和機理三統稱為非自由基機理。非自由基機理一般發生在催化劑表面有限區域,且對有機物的氧化有一定的選擇性,如金屬-有機復合物僅在有絡合能力的有機物分子(草酸等)與能形成配位化合物的過渡金屬之間產生,表面原子氧僅在氧空位產生且僅氧化被吸附的有機物。機理一為自由基機理,對有機物無選擇性,具有更強的普適性,應用前景更廣泛。常見金屬及其氧化物催化O3機理及主要催化位點總結如表1所示。 表1 污水深度處理中常見金屬及其氧化物催化O3氧化機理 近年來,活性炭、碳納米管及石墨烯等多種碳材料均用于催化O3以氧化去除有機物,且都取得了較好的效果。Snchez-Polo等[46]發現加入500 mg/L活性炭后,O3氧化去除硝基咪唑的TOC去除率提高了30%,副產物的生物毒性大幅削減,且此催化效果不受水質條件干擾,在實際水體(地表水、地下水及廢水)中也有很好的效果。Beltrn等[47]研究表明活性炭催化O3氧化草酸幾乎可實現完全礦化,并排除了單獨O3和活性炭吸附的貢獻。Gon?alves等[48]在研究碳材料催化O3氧化磺胺甲惡唑時發現,碳納米管比活性炭的催化效果更好,在礦化有機物的同時可降解反應副產物,降低中間產物的生物毒性。 活性炭是一系列無序交聯的芳香碳層狀結構,層間距多變以形成孔隙結構;理想的碳納米管是納米尺度的石墨烯柱狀結構,柱兩端以富勒烯封閉,在尾端及側壁常出現缺陷。由于結構不同,活性炭的比表面積更大,但以微孔結構為主,介孔比表面積小于碳納米管[49]。活性炭絕大多數(>90%)的表面積均為內表面,不利于O3傳質,而且在強氧化環境下缺乏結構穩定性,O3氧化后會導致比表面積和介孔結構受損。與活性炭不同的是,碳納米管外部比表面積更大,表面更容易改性以產生利于活性物質生成的官能團結構,且結構穩定性更強,因此,催化O3的效果比活性炭更加顯著。 圖3 碳材料催化O3氧化機理 石墨烯是一種由碳原子以sp2雜化軌道組成的、呈蜂巢晶格的二維碳納米材料,每個碳原子都貢獻一個位于pz軌道的未成鍵電子,在與平面垂直方向形成π鍵,因此具有優異的光學、電學和力學性能。原始的石墨烯催化效果較弱,只有通過雜原子摻雜、負載和表面化學改性等手段改變其化學性質,才可以實現更好的催化效果。例如,石墨烯經氧化后形成氧化石墨烯(GO),結構與石墨烯相同,但表面有豐富的含氧基團(包括環氧基團C-O-C、羥基C-OH、羰基C=O和羧基O-C=O),通過共價鍵與基面原子(C-O-C和C-OH)或邊緣(C=O和O-C=O)相連。通過熱或電化學方法將GO還原以生成表面含氧基團較少,但仍包含部分氧和晶格缺陷的還原氧化石墨烯(rGO)。Yin等[59]在GO中摻雜了N原子和P原子,改變了碳的sp2雜化形式,同時產生新的活性位點,提高O3氧化磺胺甲惡唑的速率。 以上研究同時指出,石墨烯及其改性材料的催化效果均隨著使用次數的增加而顯著下降,且再生手段無法完全恢復其催化能力。O3在衰減過程中產生的自由基可對石墨烯材料的物理性質及表面官能團產生影響,主要表現為D-OH和C=O等富電子堿性基團被氧化為O-C=O。Du等[63]對GO在O3環境下物理結構和化學性質的變化做了較為系統的研究:首先,被O3氧化后,GO表面褶皺更多,側邊尺寸減小并且在片狀結構中產生了空洞,即GO的基面石墨結構裂解,邊緣縮減;其次,石墨碳結構中的雙鍵只能被·OH氧化形成C-OH,此后·OH和O3可繼續通過親電加成或環偶極加成反應使C-OH被氧化為C=O和O-C=O,造成GO基面芳香環的裂解;此外,在原本存在C-OH結構的碳環中,鄰位和對位碳被活化,O3通過偶極環加成反應生成C=O或O-C=O。因此,與O3接觸后,石墨烯及其改性材料的形貌結構和表面含氧官能團均發生了較大改變,O/C比例顯著升高,C=O和O-C=O增加,催化效果減弱。 表2 污水深度處理中常見碳材料催化O3氧化機理 在各類催化劑的催化效果研究中,多數著眼于難降解及痕量有機微污染物(如抗生素、內分泌干擾物等)的去除。其中,草酸分子結構簡單且O3單獨氧化效率極低,成為催化O3氧化機制研究中最受歡迎的有機物之一。通過文獻簡要比較[18,39,43,64-66],在草酸物質的量濃度為2~8 mmol/L,催化劑投加量為1~4 g/L時,MnO2、TiO2、NiO、Co3O4、FeOOH等均表現出較好的催化效果,草酸去除率可由10%~20%(單獨O3)提高至60%~90%。但由于催化O3氧化體系中變量因素較多,如有機物濃度、催化劑投加量、O3投加量、反應時間、反應器類型、反應形式(連續流試驗/序批式試驗)等,尚未能統一衡量各類催化劑的催化效果,并結合經濟效益分析等內容,形成統一的效果評價體系。 同時,目前催化劑研究和開發應用于實際工程仍有一定限制,主要存在以下問題。其一,過渡金屬及其氧化物是有效的催化劑,但目前多數研究限于粉末型催化劑的開發,包括納米材料、介孔材料等,以期通過改變結構性質來達到更好的催化效果;但此類材料在實際應用中存在如納米顆粒團聚導致的失活、沉降性能較差等問題,需要更復雜的固液分離手段,甚至存在納米材料的生物毒性和生態風險等問題。其二,新開發的催化劑大多用復雜的物理化學手段合成,如浸漬、熱處理等,尤其是負載型催化劑;但此類催化劑面臨有效催化成分流失的問題,催化劑損耗和更換價格十分昂貴。其三,多數研究著眼于難降解及痕量有機微污染物(如抗生素、內分泌干擾物等)的去除、降解途徑及其反應動力學;而催化O3的主要目的是實現有機物的完全礦化,以及將難降解污染物轉化為小分子有機酸,為后續生物處理提供可能;實際廢水成分復雜,判斷催化效果的指標應為有機物綜合指標。其四,合成催化劑實現催化作用的pH范圍較窄,有些反應條件甚至極為苛刻,如錳氧化物僅在pH值<5時才有較可觀的催化作用,鈦氧化物也是在弱酸性條件下才能發揮最大效能;而實際廢水常在pH中性范圍且緩沖能力較強,加酸滿足催化O3氧化需求后再重新中和的過程藥耗較大;且酸性條件下金屬離子的溶出顯著增加,催化劑損耗過快,出水金屬離子也需要額外控制。 因此,在理論研究的基礎上,持續明確非均相催化O3氧化的機制、開發適用于工程應用的可持續使用的催化劑、研究其催化機理具有重要的科學意義和實踐意義。針對以下問題的研究,有助于在提升催化O3氧化效果的同時,打通科學研究到實踐應用的壁壘:(1)簡化催化劑合成及改性手段,實現溫和條件下的催化劑制備;(2)開發具有填料性質的催化劑,保證足夠的比表面積和催化位點,同時避免復雜的分離手段等問題;(3)開發性質更穩定或具有自我修復功能的催化劑,避免有效成分溶解、脫落造成的催化劑失活等問題;(4)持續研究催化O3技術與生化處理的耦合,充分發揮催化O3降解大分子有機物及生化處理礦化小分子有機物的能力,完善廢水處理鏈條。
1.3 鈰氧化物
1.4 鈷氧化物
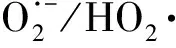
1.5 鎳氧化物
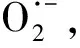
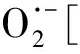
1.6 鈦氧化物
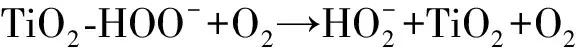
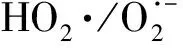

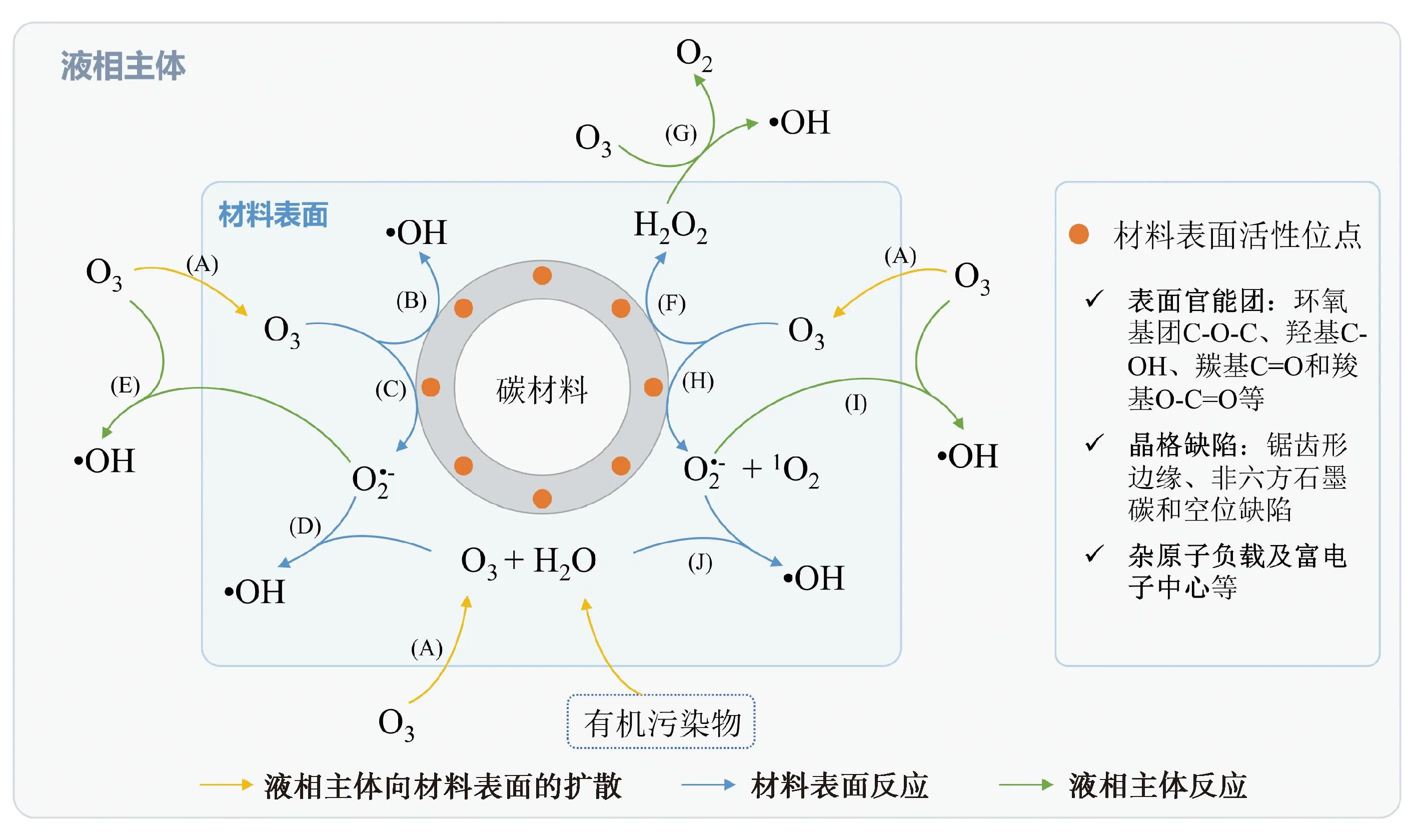
2 碳材料
2.1 活性炭和碳納米管
2.2 石墨烯
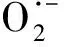

3 結論與展望
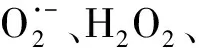
猜你喜歡
中老年保健(2021年12期)2021-11-30 02:58:01
攝影之友(影像視覺)(2019年2期)2019-03-05 08:27:14
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
中華詩詞(2018年11期)2018-03-26 06:41:34
Coco薇(2016年8期)2016-10-09 02:11:50
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
中國醫藥科學(2015年19期)2015-02-27 12:33:11
