ODC1基因對前列腺癌細胞PC-3 LncRNA表達影響的初步觀察與分析
2024-05-07 02:16:18宋尖卓
新疆醫科大學學報 2024年4期
宋尖卓, 馬 健, 陳 鵬
(新疆醫科大學附屬腫瘤醫院泌尿科, 烏魯木齊 830011)
轉錄因子鳥氨酸脫羧酶1(ODC1)是多胺生物合成中的限速酶,通過將鳥氨酸脫羧轉化為腐胺,有助于生產多胺(包括腐胺、亞精胺、精胺等)[1]。多胺是活細胞的關鍵組成部分,在細胞增殖和凋亡、衰老和生殖的調節中發揮多種作用[2]。ODC1在多種腫瘤的增生性組織中表達上調,在轉錄因子MYC基因擴增的腫瘤中發生失調,例如神經母細胞瘤、胃癌、前列腺癌和結直腸癌[3-5]。已有研究表明,ODC1基因在前列腺癌中高表達,并與臨床前列腺特異性抗原(PSA)表達水平相關,癌腺體周圍良性上皮中ODC1的表達水平隨著PSA的增加呈對數降低,ODC1抑制劑可選擇性阻斷睪酮對前列腺腫瘤發展的促進作用[6-8]。對ODC1影響調控前列腺癌的機制仍需進一步探索,本實驗擬通過高通量測序等方法和生物信息數據分析,從其對中長鏈非編碼RNA(lncRNA)表達影響的角度進行初步觀察和探索,以期為進一步研究其作用及調控機制提供一些實驗基礎和線索。
1 材料與方法
1.1 材料與試劑人前列腺癌細胞(PC-3)(普諾賽公司)。試劑包括:Ham′s F-12K(Procell公司)、0.25%胰酶(Gibco公司)、lipofectamine max(Invitrogen公司)、DMSO(MP公司)、質粒小提試劑盒(Omega公司)、胎牛血清(Gibco公司)等。
1.2 方法
1.2.1 構建細胞模型 實驗組采用三種敲低序列降低實驗誤差,分別采用siRNA ODC1-homo-1006(編號si-1)5′-GGUUGGUUUCAGCAUGUAUTT-3′(正義鏈)、5′-ACGUGACACGUUCGGAGAATT-3′(反義鏈), ODC1-homo-1156(編號si-2)5′-GCCCGGCAGAUACUAUGUUTT-3′(正義鏈)、5′- AACAUAGUAUCUGCCGGGCTT -3′(反義鏈),ODC1-homo-1440(編號si-3)5′-GCUGUGACCUGCCUG-AAAUTT-3′(正義鏈)、5′-AUUUCAGGCAGGUCACAGCTT-3′(反義鏈)進行細胞轉染,得到基因敲低組Si-1、Si-2 和Si-3。對照組NC采用siRNA siNC 5′-UUCUCCGAACGUGUCACGUTT-3′(正義鏈)、5′-ACGUGACACGUUCGGAGAATT-3′(反義鏈)進行細胞轉染。實驗組和對照組細胞樣品轉染siRNA用量為160 pmol/孔(12孔板)、Lipo 4 μL/孔、無血清培養基(μL/孔)50×2。轉染具體如下:(1)取160 pmol轉染量siRNA,加入50×2 μL稀釋體積的無血清培養基,孵育5 min;(2)同時取4 μL LipofectamineTMmax Transfection Reagent,加入無血清培養基,孵育5 min;(3)將步驟(1)加入到步驟(2)中,孵育20 min;(4)將上述混合物均勻加入細胞培養液中,置于5% CO2,37℃培養箱培養6 h,隨后更換對應的新鮮完全培養基,置于5% CO2,37℃培養箱繼續培養至48 h,收集樣品。NC組和Si組均生物學重復6次,對照組采用無關序列轉染篩選出NC-1、NC-2、NC-3,實驗組采用上述三種敲低序列轉染篩選出Si 1-1、Si 1-2、Si 1-3,Si 2-1、Si 2-2、Si 2-3,Si 3-1、Si 3-2、Si 3-3。
1.2.2 驗證細胞模型 Trizol法提取上述轉染48 h的樣品細胞RNA,利用引物Hum GAPDH 5′-GGTCGGAGTCAACGGATTTG-3′(正義鏈)、5′-GGAAGATGGTGATGGGATTTC-3′(反義鏈)和引物ODC1 5′-CCGAAGTAGAGGAACAGGAT-3′(正義鏈)、5′-TTAATACTAGCCGAAGCACAG-3′(反義鏈),進行qRT-PCR檢測分析不同樣本之間的相對表達量,檢測ODC1基因在mRNA水平上的表達量。WB檢測ODC1蛋白在敲低ODC1基因的PC-3細胞模型中的表達情況。取1 μg檢測合格的總RNA進行RNA-seq文庫的制備。RNA-seq文庫制備后使用Illumina Novaseq 6000(武漢瑞興生物科技有限公司),采用PE150模式進行高通量測序。在獲得原始序列之后,去掉測序中低質量的片段。使用FastQC(版本0.9.5)對fastq格式的質量過濾后片段進行質量檢測。然后,用TopHat2軟件把經過篩選的序列比對到人GRCH38基因組上面,容許不超過4個堿基錯配。刪除能夠與多個基因組位置匹配的序列,僅保留具有唯一基因組位置的序列,進行后續分析。
1.2.3 統計處理與生信分析 采用SPSS18.0軟件處理數據,計量資料兩組間比較采用兩獨立樣本t檢驗,各組之間差異采用單因素方差分析,實驗組和對照組中lncRNA表達量變化采用Fisher檢驗,以P<0.05為差異有統計學意義。
采用目前應用最廣泛的編碼潛能分析方法(CPC分析、CNCI分析、CPAT分析、LGC分析)對候選lncRNA進行判定篩選,使用DESeq2軟件對篩選基因進行差異表達分析,然后估計基因離散度,再擬合負二項分布模型,采用Wald或似然比假設檢驗。檢驗標準為表達變化的絕對比值fold change(FC≥2 or ≤1/2)和假設幾率(P<0.05),篩選出表達顯著上調基因和表達顯著下調基因,作圖分析軟件采用Cluster3.0。對所有差異表達的lncRNA和所有差異表達的基因進行共表達分析,同時計算差異表達的lncRNA表達基因之間的皮爾森相關系數,篩選滿足相關系數絕對值>0.6且P<0.01的關系對。得到差異基因后,分析lncRNA順式作用靶標,再對靶標進行GO分類富集性分析和KEGG分析解析其功能。首先綜合GO和KEGG數據庫資源向各個通路映射,統計每個通路中的基因數,然后用超幾何分布檢驗,以全基因組的GO和KEGG注釋情況為背景,得到待分析基因顯著富集的通路。最終展示排名前十的通路。
1.2.4 基因驗證 qRT-PCR檢測驗證上述實驗中各樣本lncRNA的表達量改變。針對基因序列設計引物LINC00973-F 5′-GGCTTCATCAATAAGGTATTCC-3′、LINC00973-R 5′-GAATAATTGTCCTTGCCTCAGA-3′。TERC-F 5′-CTAGAATGAA-CGGTGGAAGGC-3′、TERC-R 5′-TAACTGAGA-AGGGCGTAGGC-3′。LINC00638-F 5′- GACCC-GTCCCTTTGAGGAT-3′、LINC00638-R 5′-AGCGA-GGATGGTGTCTGAG-3′。具體采用反轉錄試劑盒(R323-01,諾唯贊,中國),在thermocycle儀器(T100,Bio-Rad,USA)上進行cDNA合成,反轉錄條件為42℃,5 min,37℃,15 min,85℃,5 s。然后,在ABI QuantStudio 5儀器上進行q-PCR反應,設置的程序為95℃預變性10 min,95℃變性15 s,60℃退火延伸1 min,變性和延伸執行40個循環。每個樣品進行3次技術重復。接下來計算每個轉錄本mRNA的相對表達量,使用GAPDH作為內參基因進行標準化,采用2- ΔΔCT方法進行分析[9]。
2 結果
2.1 細胞模型構建及ODC1基因敲低后lncRNAs差異分析
2.1.1 qRT-PCR結果 NC組mRNA表達量分別為1、0.793、1.03。實驗組Si-1組mRNA表達量分別為0.112、0.113、0.123。實驗組Si-2組mRNA表達量分別為0.079、0.106、0.124。實驗組Si-3組mRNA表達量為0.112、0.112、0.13。各組取平均值后mRNA表達量分別為NC組0.941,Si-1組0.116,Si-2組0.103,Si-3組0.118。與NC組相比,Si組mRNA表達水平顯著降低。WB結果顯示,ODC1分子量為51 KDa,實際檢測分子量為50 KDa,在70 KDa存在很亮的雜帶;從WB圖條帶來看,NC組與Si組相比,NC組有條帶,Si組無條帶,敲低效率較高,見圖1。從mRNA和蛋白水平提示PC3敲低ODC1成功。
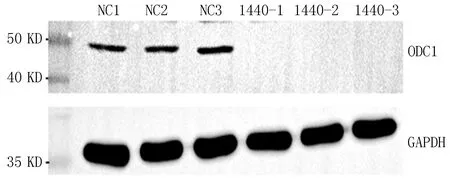
圖1 ODC1在實驗組和對照組中的表達
2.1.2 lncRNA預測 對RNA-seq數據進行分組,使用StringTie對各組數據進行組裝并預測轉錄本,CPC、CNCI、CPAT、LGC這4種分析方法取交集后篩選的新預測lncRNA有341個,結果見圖2。
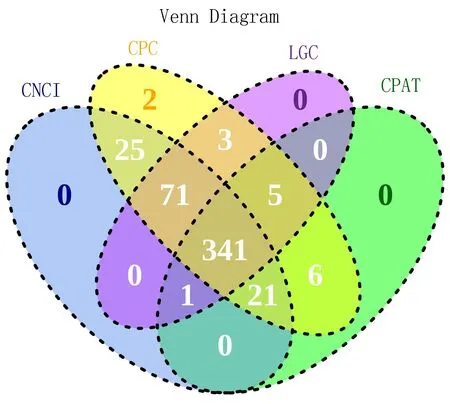
注: 藍色部分表示采用編碼潛能分析方法CNCI篩選出的lncRNA數量;黃色表示采用CPC分析方法篩選出的lncRNA數量;紫色表示采用LGC分析方法篩選出的lncRNA;綠色表示采用CPAT分析方法篩選出的lncRNA。圓圈重疊的部分表示2種及2種以上分析方法篩選出的共有的非編碼RNA數量。
2.1.3 實驗組與對照組細胞間lncRNA差異分析 采用標準DElncRNA比較不同樣本,獲得差異表達lncRNA。實驗組顯著上調表達lncRNA有154個,前10的有LINC00638、LINC00871、LINC02714、LINC00992、LINC01252、LINC00461、LINC01238、LINC01812、LINC01844、LINC00957等。下調表達的lncRNA有128個,前10的有TERC、LINC00973、LINC00592、LINC02454、LINC01444、LINC00997、LINC02085、LINC01089、LINC01285、LINC02029;差異lncRNA聚類分析揭示實驗組與對照組對比鮮明,樣本一致性好,見圖3、4。
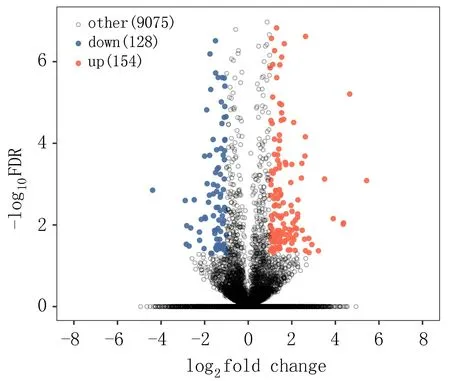
圖3 lncRNA差異分析火山圖
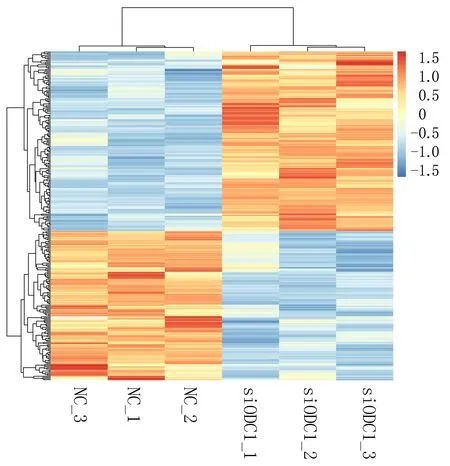
圖4 差異表達lncRNA熱圖
2.1.4 lncRNA順式調控靶標分析 在反式調控的關系對中將co-location的閾值設定為lncRNA上下游100 kb,后續通過對co-location的lncRNA和mRNA之間計算皮爾森相關系數進行共表達(co-expression)分析,篩選滿足相關系數絕對值>0.6且P<0.01的lncRNA-target關系對,再對co-location和co-expression兩個數據集取交集,得到lncRNA的順式作用靶標。lncRNA與所有表達基因的共表達順式調控靶標數目789個,差異lncRNA與所有表達基因的順式調控靶標數目為142個,差異lncRNA與差異表達基因共表達的順式調控靶標數目為38個,結果見表1。

表1 lncRNA的順式作用靶標數目統計表
2.2 lncRNA靶標GO和KEGG Pathway富集分析通過基因功能富集分析、生物學過程富集分析和KEGG基因通路富集分析,lncRNA順式調控靶標基因參與磷脂酰肌醇3激酶/蛋白激酶B信號通路(PI3K-Akt信號通路)、Ras等信號通路并與腫瘤血管生成、細胞群增殖和正調控細胞分裂等密切相關。如LINC00638、PRLR、NRAS、PDGFRB等參與PI3K-Akt信號通路及Ras信號通路;TERC、CDKN1A、CASP3、LINC00973等也與腫瘤信號通路相關。這些生物學功能表明lncRNA順式靶標基因與腫瘤形成進展等聯系密切,見圖5~7。
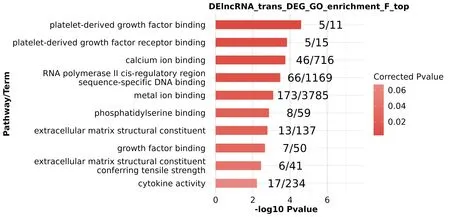
圖5 lncRNA靶標GO功能(F)富集分析結果、基因功能及結合受體情況

圖6 lncRNA靶標GO生物學過程(P)富集分析結果

圖7 lncRNA靶標KEGG Pathway基因通路富集分析
2.3 驗證GO和KEGG富集分析得到的功能lncRNA基因進行LncRNA順式調控靶標GO和KEGG通路富集分析后,得到LncRNA LINC00973、TERC、LINC00638。
補充驗證實驗,結果顯示,與對照組相比,實驗組Si-1、Si-2、Si-3中LINC00973和TERC的表達量降低,差異有統計學意義(P<0.000 1);與對照組相比,實驗組LINC00638表達量升高,差異有統計學意義,見表2。q-PCR結果顯示,LINC00973、TERC表達顯著下調、LINC00638表達顯著上調,與GO和KEGG富集分析得到的lncRNA差異表達情況一致,見表3。
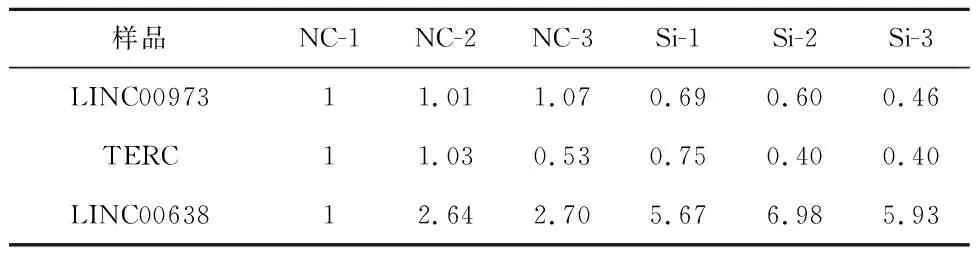
表2 實驗組和對照組中LINC00973、TERC和LINC00638表達的比較
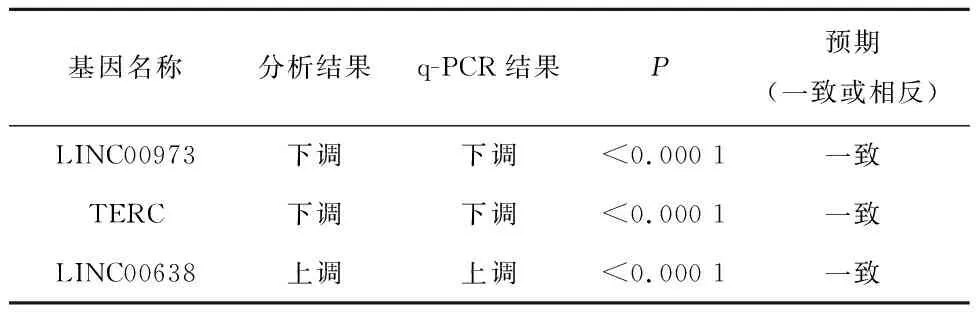
表3 q-PCR結果展示
3 討論
ODC1基因在前列腺癌細胞中顯著高表達,且與患者預后相關[6]。ODC1影響調控前列腺癌的機制可能與雄激素相關通路有關,但詳細機制仍需進一步探索,本實驗通過高通量測序方法探索ODC1基因對中長鏈非編碼RNA(lncRNA)表達的影響,以期從這一角度為研究其影響調控前列腺癌的機制提供一些可能的依據。我們在體外實驗PC-3細胞中敲低ODC1后,觀察到眾多lncRNAs基因隨之差異表達,選擇部分文獻已有報道與腫瘤相關、特別是和前列腺癌相關的lncRNAs進行了驗證,結果顯示,與LncRNA順式調控靶標GO和KEGG通路富集分析預期結果一致,提示從這一角度繼續深入研究是可行的。
lncRNAs不同于編碼蛋白基因,當受到干擾時,lncRNAs的錯誤調控會導致基因組應激[10],已有研究表明,lncRNAs與包括前列腺癌在內的多種人類惡性腫瘤的發生和發展有關[11-12]。
本實驗中驗證的lncRNA TERC、LINC00973、LINC00638,在既往的研究中發現具有促進腫瘤細胞增殖及侵襲的作用[13],本實驗富集通路結果顯示,這三個lncRNA與多個腫瘤信號通路相關。TERC基因在端粒酶依賴的端粒延伸和維持中起著重要作用[13],文獻表明,TERC突變可以激活前列腺癌細胞LAPC4的端粒延長代替機制,并導致癌細胞繼續長期增殖[14]。有研究報道,LINC00973在結腸癌、腎透明細胞癌及非小細胞肺癌中高表達[15-17],它通過結合糖酵解關鍵酶LDHA(乳酸脫氫酶A)并增強酶活性,進而加速腫瘤細胞的有氧糖酵解,最終促進腫瘤細胞增殖,敲低該RNA的表達,能夠抑制多種腫瘤細胞的增殖[18],根據本研究結果可以推測,LINC00973的高表達也是通過加速了前列腺腫瘤細胞有氧糖酵解過程,以此促進前列腺癌的進展。還有研究顯示,非小細胞肺癌中過表達LINC00638,通過調控IRS1/磷酸肌醇3-激酶(PI3K)/Akt信號通路促進腫瘤增殖、生長、遷移和侵襲,抑制非小細胞肺癌細胞的凋亡,miR-541-3p可能是LINC00638的潛在靶點[19],在肝細胞癌中,LINC00638參與LINC00638/miR-4732-3p/ULBP1軸,通過PD-L1促進肝細胞癌的免疫逃逸,導致肝細胞癌進展[20],在前列腺癌中LINC00638是否也是參與PI3K/Akt信號通路而促進腫瘤進展也需要實驗進一步證明。
本實驗只挑選了以上三個已知重要的lncRNA進行驗證,可能對全面解釋調控機制來說不夠充分。另外,本實驗是采用人前列腺癌PC-3細胞作為研究基礎的,后續我們還將通過體內實驗來驗證ODC1高表達是否足以驅動前列腺癌進展。再者,lncRNA與其他物質結合形成的復合物是否參與調控后續生物學進程,還需要進一步通過諸如RNA pull-down、ChlRP、RIP、CHIP-s等技術對候選lncRNA的作用機制作進一步研究,這也是我們后續深入研究的方向。
本次的研究結果提示了ODC1基因通過調控致癌lncRNAs參與前列腺癌的發生或進展的新線索,及其作為治療干預靶點的潛在意義,為進一步研究ODC1基因在前列腺癌中的作用及調控機制提供了一定的實驗基礎。
猜你喜歡
音樂探索(2022年2期)2022-05-30 21:01:37
民用飛機設計與研究(2020年4期)2021-01-21 09:15:02
家庭醫學(下半月)(2020年3期)2020-05-30 12:42:02
家庭醫學(下半月)(2020年3期)2020-05-30 12:42:00
家庭醫學(下半月)(2020年3期)2020-05-30 12:42:00
小天使·一年級語數英綜合(2019年8期)2019-08-27 02:23:00
中國生殖健康(2019年7期)2019-01-06 09:27:34
電子制作(2018年18期)2018-11-14 01:48:24
小學科學(學生版)(2018年7期)2018-08-13 09:33:04
山東工業技術(2016年15期)2016-12-01 05:31:22
