染色質(zhì)調(diào)節(jié)因子對(duì)乳腺癌預(yù)后的預(yù)測(cè)價(jià)值
2024-05-07 10:07:58凌鏗金麗琴
浙江醫(yī)學(xué) 2024年8期
凌鏗 金麗琴
乳腺癌在2020 年的全球發(fā)病例數(shù)高達(dá)226 萬例,已超越肺癌的220 萬例,成為全球發(fā)病率第一的癌癥[1-2]。盡管醫(yī)學(xué)技術(shù)不斷進(jìn)步,如靶向治療和免疫治療等在臨床的廣泛應(yīng)用,但乳腺癌患者的總體生存率仍不盡如人意[3-4]。近年研究顯示,多基因標(biāo)記有助于乳腺癌的風(fēng)險(xiǎn)分層和預(yù)后預(yù)測(cè)[5]。染色質(zhì)調(diào)節(jié)因子(chromatin regulators,CRs)在腫瘤中的表觀遺傳學(xué)改變被視為關(guān)鍵標(biāo)志[6-7],其失調(diào)可能導(dǎo)致癌癥等多種疾病的發(fā)生。然而,關(guān)于CRs 與乳腺癌之間關(guān)系的系統(tǒng)研究仍然較少。本研究通過生物信息學(xué)方法,探討CRs 在乳腺癌中的表達(dá)模式及其預(yù)測(cè)預(yù)后的潛在價(jià)值,旨在了解CRs 的作用機(jī)制并尋找新的治療靶點(diǎn)。
1 材料和方法
1.1 差異表達(dá)CRs 的數(shù)據(jù)收集和識(shí)別 從公共數(shù)據(jù)庫癌癥基因組圖譜(The Cancer Genome Atlas,TCGA,https://portal.gdc.cancer.gov)獲取111 個(gè)正常乳腺組織和1 057 個(gè)乳腺癌組織的mRNA 表達(dá)和相關(guān)臨床信息,以及從前期專題研究中檢索到870 個(gè)CRs[8]。這些mRNA 采用R 軟件preprocess Core 軟件中的normalize.quantiles 函數(shù)進(jìn)行數(shù)據(jù)標(biāo)準(zhǔn)化處理,根據(jù)相應(yīng)平臺(tái)注釋信息,統(tǒng)計(jì)將探針I(yè)D轉(zhuǎn)換為gene symbol。根據(jù)|logFC|>1 和假發(fā)現(xiàn)率(false discovery rate,F(xiàn)DR)<0.05 的標(biāo)準(zhǔn),使用基于R軟件的limma軟件識(shí)別差異表達(dá)的CRs。
1.2 基于CRs 的預(yù)測(cè)模型的構(gòu)建與驗(yàn)證 采用單因素Cox 回歸分析對(duì)所有候選CRs進(jìn)行初步篩選,從而確定與乳腺癌患者總體生存率顯著相關(guān)的CRs。隨后,采用最小絕對(duì)值收斂和選擇算子算法(least absolute shrinkage and selection operator,LASSO)-Cox 回歸分析法中的交叉驗(yàn)證方法確定最佳的懲罰參數(shù)λ值,以最終確定包含在預(yù)后風(fēng)險(xiǎn)模型中的CRs。LASSO-Cox 回歸不僅能夠處理變量選擇問題,還能有效地控制模型的復(fù)雜度,減少過擬合的風(fēng)險(xiǎn),為乳腺癌患者提供一個(gè)可靠的預(yù)后評(píng)估工具。然后計(jì)算每例乳腺癌患者的風(fēng)險(xiǎn)分?jǐn)?shù)(Risk score),Risk score=(Coef 1×expression mRNA 1)+(Coef 2×expression mRNA 2)+…+(Coef n×expression mRNA n),其中Coef 是相應(yīng)mRNA 的LASSO-Cox 回歸模型系數(shù)。以所有患者Risk score 的中位數(shù)將乳腺癌患者分為高風(fēng)險(xiǎn)組和低風(fēng)險(xiǎn)組。采用Kaplan-Meier 法進(jìn)行生存分析,評(píng)估兩組患者的預(yù)后。使用survival ROC 軟件繪制時(shí)間依賴性ROC 曲線評(píng)估風(fēng)險(xiǎn)模型的預(yù)測(cè)效能,計(jì)算AUC,數(shù)值越接近1,表示模型的預(yù)測(cè)效能越強(qiáng)。
1.3 基于Risk score 和臨床變量構(gòu)建列線圖 將模型與乳腺癌患者其他臨床變量相結(jié)合以提高預(yù)測(cè)準(zhǔn)確性,其中臨床變量包括患者的年齡、癌癥分期(StageⅠ、Ⅱ、Ⅲ、Ⅳ期)、腫瘤大小(T 分類)、淋巴結(jié)轉(zhuǎn)移情況(N 分類)、遠(yuǎn)處轉(zhuǎn)移情況(M 分類)。采用單因素Cox 回歸分析以評(píng)估Risk score 以及上述臨床變量對(duì)乳腺癌患者預(yù)后的影響,隨后采用多因素Cox 回歸分析確定Risk score 對(duì)乳腺癌患者預(yù)后的獨(dú)立預(yù)測(cè)價(jià)值。應(yīng)用上述臨床變量和基于CRs 的特征Risk score 構(gòu)建列線圖,以評(píng)估乳腺癌患者1、3、5 年總生存期(overall survival,OS)。采用校準(zhǔn)曲線來評(píng)估列線圖的預(yù)測(cè)效能。
1.4 統(tǒng)計(jì)學(xué)處理 采用R 4.2.1 統(tǒng)計(jì)軟件。組間CRs表達(dá)水平的比較采用Wilcoxon 秩和檢驗(yàn),高風(fēng)險(xiǎn)組和低風(fēng)險(xiǎn)組生存時(shí)間的比較采用log-rank 檢驗(yàn)。P<0.05 為差異有統(tǒng)計(jì)學(xué)意義。
2 結(jié)果
2.1 基于CRs 的模型建立和驗(yàn)證 與正常乳腺組織相比,TCGA-乳腺癌數(shù)據(jù)庫中有127 個(gè)差異表達(dá)的CRs,見圖1(插頁)。根據(jù)P值排序,前20 個(gè)具有預(yù)后價(jià)值的基因見圖2。然后,使用LASSO-Cox 回歸分析篩選出16 個(gè)差異CRs 并構(gòu)建乳腺癌患者預(yù)后預(yù)測(cè)能力的特征(ACTL6B+ASCL1+CHEK1+FBXL19+FOXA1+HMGB3+IDH2+MAZ+MECOM+RAD54B+SMYD3+SP140+TDRD12+TDRKH+TONSL+UBE2T),成功構(gòu)建了風(fēng)險(xiǎn)模型。根據(jù)16 個(gè)CRs 的相關(guān)系數(shù)計(jì)算Risk score,Risk score=(0.023 5×ACTL6B 表達(dá))+(0.003 0×ASCL1 表達(dá))+(0.003 4×CHEK1 表達(dá))+(0.019 3×FBXL19 表達(dá))+(0.001 5×FOXA1 表達(dá))+(0.002 0×HMGB3 表達(dá))+(0.001 1×IDH2 表達(dá))+(-0.010 3×MAZ 表達(dá))+(0.059 8×MECOM 表達(dá))+(0.075 0×RAD54B 表達(dá))+(0.007 1×SMYD3 表達(dá))+(-0.097 8×SP140 表達(dá))+(0.018 4×TDRD12 表達(dá))+(0.012 2×TDRKH 表達(dá))+(0.008 3×TONSL 表達(dá))+(0.002 3×UBE2T 表達(dá))。Kaplan-Meier 生存曲線分析顯示,高風(fēng)險(xiǎn)組死亡率明顯高于低風(fēng)險(xiǎn)組(P<0.001);時(shí)間依賴性ROC 曲線分析顯示,TCGA 數(shù)據(jù)庫中基于CRs 的特征在5 年時(shí)的AUC 為0.778,見圖3(插頁)。利用熱圖顯示高風(fēng)險(xiǎn)組和低風(fēng)險(xiǎn)組之間16 種CRs 的差異,見圖4(插頁)。
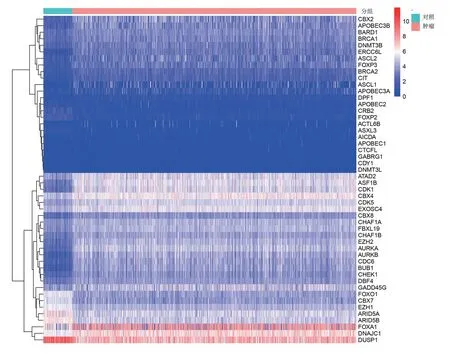
圖1 乳腺癌前50 個(gè)差異表達(dá)CRs 的熱圖
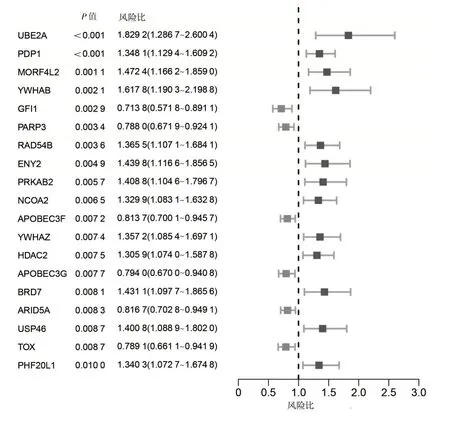
圖2 前20 個(gè)預(yù)后基因的森林圖
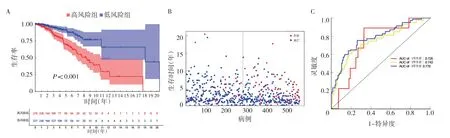
圖3 TCGA-乳腺癌數(shù)據(jù)集基于預(yù)后CRs 的模型分析(A:Kaplan-Meier 高風(fēng)險(xiǎn)與低風(fēng)險(xiǎn)組乳腺癌患者生存分析;B:基于中位Risk score 的生存狀態(tài)分布;C:總生存期Risk score 的時(shí)間依賴性ROC 曲線)
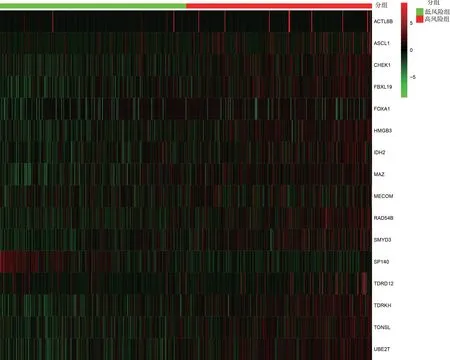
圖4 高風(fēng)險(xiǎn)組和低風(fēng)險(xiǎn)組之間16 種CRs 差異的熱圖
2.2 基于CRs 特征構(gòu)建的風(fēng)險(xiǎn)模型與乳腺癌患者臨床特征之間的相關(guān)性分析 結(jié)果顯示,該風(fēng)險(xiǎn)模型在預(yù)測(cè)高風(fēng)險(xiǎn)組和低風(fēng)險(xiǎn)組乳腺癌患者不同臨床特征中的預(yù)后方面具有明顯差異,相關(guān)性熱圖見圖5(插頁)。Kaplan-Meier 生存曲線分析得出,低風(fēng)險(xiǎn)組患者中年齡≤65 歲、StageⅠ~Ⅱ期、Ⅲ~Ⅳ期、T0~T1、T2~T3、N0~N1、N2~N3者OS 均高于高風(fēng)險(xiǎn)組患者(均P<0.05),但兩組患者中年齡>65 歲者OS 比較差異無統(tǒng)計(jì)學(xué)意義(P=0.170),見圖6。
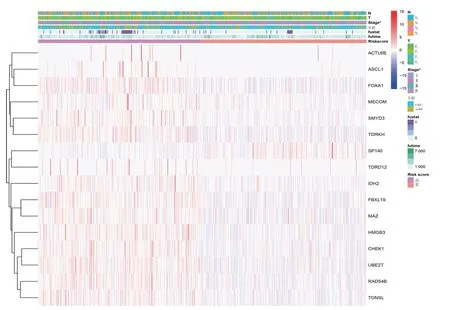
圖5 基于CRs 構(gòu)建的預(yù)后Risk score 與乳腺癌患者的不同臨床特征之間相關(guān)性分析的熱圖
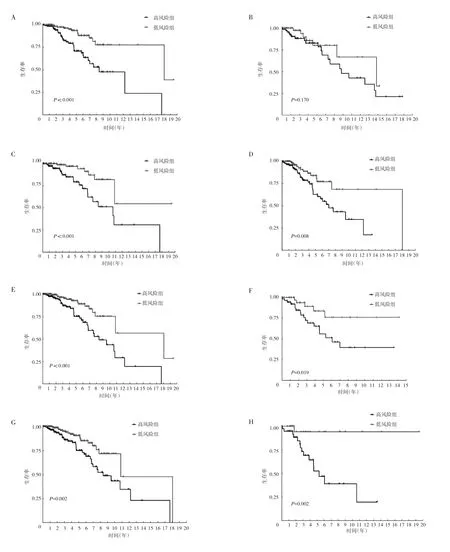
圖6 不同臨床特征高風(fēng)險(xiǎn)組和低風(fēng)險(xiǎn)組患者的Kaplan-Meier 生存曲線(A:年齡≤65 歲;B:年齡>65 歲;C:N0~N1期;D:N2~N3期;E:Stage Ⅰ~Ⅱ期;F:Stage Ⅲ~Ⅳ期;G:T0~T1期;H:T2~T3期)
2.3 預(yù)測(cè)乳腺癌患者生存率的列線圖 結(jié)合Risk score 以及其他臨床變量(患者年齡、癌癥分期、T 分類和N 分類)構(gòu)建的列線圖見圖7A。為驗(yàn)證該預(yù)測(cè)模型的可行性和準(zhǔn)確性,進(jìn)一步采用校準(zhǔn)曲線,結(jié)果表明預(yù)測(cè)模型在1、3 和5 年生存率的預(yù)測(cè)上與實(shí)際觀察到的生存率高度一致,見圖7B。
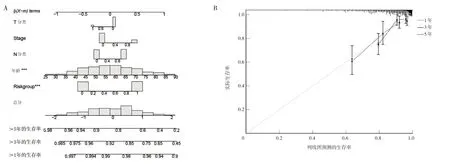
圖7 預(yù)測(cè)乳腺癌患者生存率的列線圖與校準(zhǔn)曲線(A:基于多因素的乳腺癌患者1、3 和5 年的生存率預(yù)測(cè)的列線圖;***表示這兩個(gè)變量在Cox 比例風(fēng)險(xiǎn)模型中對(duì)于預(yù)測(cè)結(jié)果有非常顯著的影響;B:列線圖的校準(zhǔn)曲線圖)
2.4 相關(guān)基因在腫瘤組織與正常乳腺組織中的基因表達(dá)差異采用limma 軟件統(tǒng)計(jì)分析乳腺癌組織與正常乳腺組織之間的基因表達(dá)差異,識(shí)別在疾病進(jìn)程中顯著上調(diào)或下調(diào)的關(guān)鍵CRs,其中ACTL6B、ASCL1、CHEK1、FBXL19、FOXA1、HMGB3、IDH2、MAZ、RAD54B、SMYD3、TDRD12、TDRKH、TONSL、UBE2T 在腫瘤組中顯著高表達(dá),而MECOM 在腫瘤組中顯著低表達(dá)。
3 討論
乳腺癌是全球最常見的女性惡性腫瘤,其復(fù)雜性和對(duì)其發(fā)生及進(jìn)展的有限了解使得其預(yù)后和治療具有挑戰(zhàn)性。盡管CRs 在乳腺癌的發(fā)生中已被證明具有多種功能,但對(duì)其在乳腺癌中的臨床意義的綜合分析仍然很少。
本研究通過利用生物信息學(xué)工具,從TCGA 數(shù)據(jù)庫中篩選出了127 個(gè)在乳腺癌組織和正常乳腺組織之間差異表達(dá)的CRs。這些CRs 的差異表達(dá)可能與乳腺癌的發(fā)病機(jī)制、預(yù)后和治療有關(guān)。隨后,進(jìn)一步分析確定了16 個(gè)與乳腺癌預(yù)后顯著相關(guān)的CRs,并基于這些CRs 構(gòu)建了風(fēng)險(xiǎn)模型。該模型的生存率和ROC 曲線分析均顯示出良好的預(yù)測(cè)效能,為臨床醫(yī)師提供了一個(gè)新的工具來評(píng)估乳腺癌患者的預(yù)后。
對(duì)于這些關(guān)鍵的CRs,如ACTL6B、ASCL1、CHEK1和FBXL19 等,它們?cè)谀[瘤中的作用已被廣泛研究[8-11]。例如,ACTL6B 在染色質(zhì)重塑和組蛋白乙酰化中起到關(guān)鍵作用[12],而CHEK1 則被認(rèn)為是乳腺癌的潛在預(yù)后和治療靶點(diǎn)[13]。這些發(fā)現(xiàn)不僅增強(qiáng)了對(duì)乳腺癌發(fā)病機(jī)制的了解,還為未來的治療策略提供了新的方向。
然而,本研究也存在一些局限性,如CRs 如何調(diào)控乳腺癌細(xì)胞的生物學(xué)行為仍需要進(jìn)一步的實(shí)驗(yàn)驗(yàn)證。此外,為了驗(yàn)證預(yù)后模型的實(shí)用性,還需要在多中心臨床隊(duì)列中進(jìn)行進(jìn)一步的研究。
綜上所述,本研究為了解乳腺癌的發(fā)病機(jī)制、預(yù)測(cè)預(yù)后以及發(fā)現(xiàn)新的治療靶點(diǎn)提供了重要的線索。但為了將這些發(fā)現(xiàn)應(yīng)用于臨床實(shí)踐,還需要進(jìn)一步的研究和驗(yàn)證。希望未來的研究能夠基于這些初步發(fā)現(xiàn),進(jìn)一步深化對(duì)乳腺癌的理解,為患者帶來更好的治療效果和生活質(zhì)量。
猜你喜歡
童話王國·奇妙邏輯推理(2024年5期)2024-06-19 16:03:38
中老年保健(2022年6期)2022-08-19 01:41:48
音樂探索(2022年2期)2022-05-30 21:01:37
中學(xué)生數(shù)理化·七年級(jí)數(shù)學(xué)人教版(2020年10期)2020-11-26 08:24:50
數(shù)學(xué)物理學(xué)報(bào)(2020年2期)2020-06-02 11:29:24
小天使·一年級(jí)語數(shù)英綜合(2019年8期)2019-08-27 02:23:00
中國生殖健康(2019年2期)2019-08-23 08:11:42
中國生殖健康(2019年6期)2019-01-06 09:20:12
小學(xué)科學(xué)(學(xué)生版)(2018年7期)2018-08-13 09:33:04
祝您健康(2018年5期)2018-05-16 17:10:16
