DFT法研究三聚甲醛開環聚合熱力學
2024-05-08 13:27:28李增昌陸全華劉光臻
山東化工 2024年7期
李增昌,陸全華,劉光臻
(百色學院 化學與環境工程學院 廣西城市水環境重點實驗室,廣西 百色 533000)
開環聚合是高分子化學中一個重要的領域,而環醚的開環聚合是開環聚合反應中具有代表性的一類。環醚開環聚合可用來合成多種應用廣泛的產品,因此是研究較多的一大類反應。環狀醚類化合物中,應用量最大的應該屬于聚甲醛,聚甲醛英文縮寫POM,是一種性能優良的熱塑性工程塑料。其表面光滑有光澤,且其密度大,可在溫度為-40~100 ℃的范圍內長期使用。聚甲醛雖可由甲醛直接聚合,但由甲醛直接聚合得到的產品相對分子質量通常不高,并且需要較高的技術條件。據說當今只有少數大科技公司掌握了由甲醛直接聚合得到性能優良的聚甲醛。聚甲醛實際上更多的是由三聚甲醛即1,3,5-三氧六環開環聚合得到,或是與1,3-二氧五環共聚得到[1]。另外,它的耐磨性和自潤滑性也比絕大多數工程塑料優越,且有良好的耐油性[2]。雖然聚甲醛也有不足,比如易降解、不耐酸堿等,但瑕不掩瑜,是用量僅次于聚酰胺和聚碳酸酯的第三大通用工程塑料,在很多場合得到了應用。
從環醚結構來看,單體的環張力是一個非常重要的熱力學因素,這一因素決定了其能否進行開環聚合反應。其開環聚合的可能性,可根據熱力學數據即反應前后的焓變或是自由能來進行判斷。一般說來,開環的焓變主要來自環張力的釋放。環張力大,焓變也就越大。自由能變化包括焓變和熵變兩部分。而熵變一項與熱焓相比數值較小,聚合趨勢主要取決于焓變,而焓變又包括內能的變化和體積與壓力的積兩項。即ΔE和PΔV兩項。在開環聚合過程中,PΔV的數值要遠遠小于ΔE的數值,故而內能的變化即ΔE基本上決定了開環的可行性。高分子化學的基本原理認為,開環聚合過程中的ΔE來自單體的環張力,而環張力主要是由相鄰碳上直立鍵上的氫原子電子云之間斥力造成的[3]。一般情況下,六元環醚因可成椅式構象,像四氫吡喃。1,4-二氧六環等單體都是如此,張力較小,六元環狀單體除1,3,5-三氧六環外,其他單體均不能發生聚合,正如前面所說,六元環狀單體的內能變化主要來自椅式構象或是船式構象上直立氫原子之間的斥力,而多數情況下,這些氫原子的斥力較小,所以,該領域內的主要熱點是研究三氧六環的聚合。
鑒于開環聚合在生產上的實際應用,系統地對開環聚合的熱力學和動力學研究非常重要,然而這需要投入大量的人力和物力,即便如此,有些數據也難以獲得。而理論計算,在某些方面可以彌補實際測量的不足。通過DFT法研究開環聚合熱力學,可以為開發新型高分子材料提供理論指導。具體來說,研究開環聚合反應的熱力學參數,如焓和熵,可以幫助優化聚合反應的條件,并預測聚合產物的性質。這對于合成新型功能高分子材料或改進現有材料的性能具有重要意義。因此,DFT法研究開環聚合熱力學是一項具有潛在應用價值的研究工作。密度泛函理論的最早可以追溯到1920年前后,它的基本概念最早起源于托馬斯和費米等人提出的Thomas-Fermi-Dirac模型[4],在1964年后由Kohn正式提出密度泛函理論,Kohn也因提出該理論獲得1998年諾貝爾化學獎,這是對計算化學領域的一大肯定,也顯示了理論在計算量子化學領域的核心作用。自1990年以來,DFT方法發展迅速,已經在理論計算的很多方面如鍵能、預測化合物結構和反應機理等方面取得巨大成功。這是一種應用廣泛的第一性原理計算方法,密度泛函包含了電子相關能的計算,在提升了計算速度的同時,結果也能保持較高計算精度[20]。密度泛函理論是目前量子化學中最為流行和實用的計算方法之一,且其在材料科學、物理學、化學等領域都得到了廣泛的應用。Guassian高斯量子化學計算軟件是一個功能強大的工具,其有多種計算方法。其中的密度泛函理論,是目前備受青睞的計算方法,此計算方法具有計算精度高、計算速度快的優點,可以單純通過計算來獲取實驗中無法獲得中間體證據[5-8];能單獨提出某些反應的機理,對反應現象和數據進行解釋,或是對實驗機理進行驗證并能對實驗數據做必要的補充[9-11]。本文就是利用高斯量子化學計算軟件Guassian和Chem3D,對一系列開環單體開環聚合的熱力學數據進行了計算,一方面可以用來彌補實際測量的不足,另一方面則可以回答高分子化學教學中的某些問題。
眾所周知,環狀單體的開環聚合,其動力主要來自環的張力,在許多高分子化學教科書中,均提到了1,3,5-三氧六環的結構與前者不同,但有什么不同,卻均未給出答案。從教學的角度看,也有必要對1,3,5-三氧六環的結構與開環機理進行深入研究。
1 計算方法
本文全部通過密度泛函理論(Density Functional Theory),所有分子均在B3LYP水平上應用6-311G++(d,p)基組進行計算。選取了四氫吡喃、1,3-二氧六環、1,4-二氧六環和1,3,5-三氧六環(即三聚甲醛)四種六元環作對比。環狀單體分別對椅式構象和船式構象進行優化和頻率分析得到其穩定態的最低能量。環己烷、1,3-二氧六環、1,4-二氧六環三種環狀化合物均是椅式構象能量較低,故取椅式構象的能量作為其單體的開環前單體的能量。1,3,5-三氧六環船式構象能量較低,故取其船式構象作為此單體的能量。由于Gaussian不適合對聚合物的片段進行直接計算,需用間接法求得。其中,環己烷開環產物的能量,由完全鋸齒狀正十二烷和完全鋸齒狀正己烷的能量差求得,1,3-二氧六環、1,4-二氧六環由鋸齒狀的含有端基的二聚體和含有端基的鏈狀單體能量差求得。1,3,5-三氧六環的能量由帶端基的螺旋形構象的二聚體和含有端基的單體之差求得。
2 結果與討論

六元環及開鏈單元結構數據如表1所示。
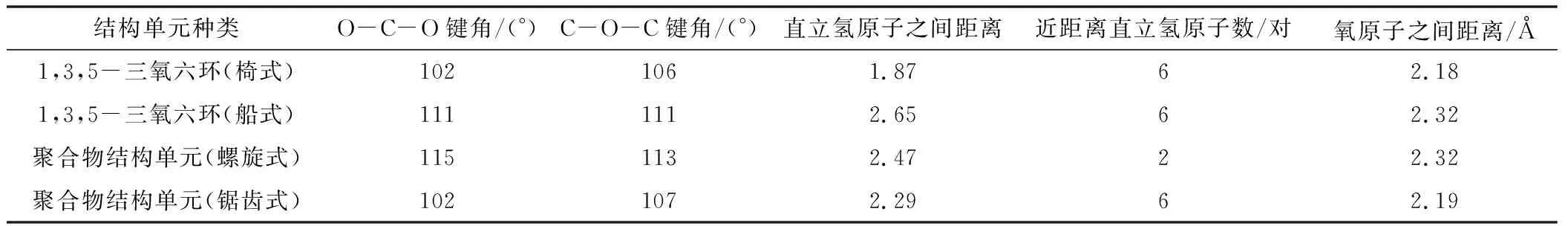
表1 六元環及開鏈單元結構數據
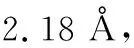
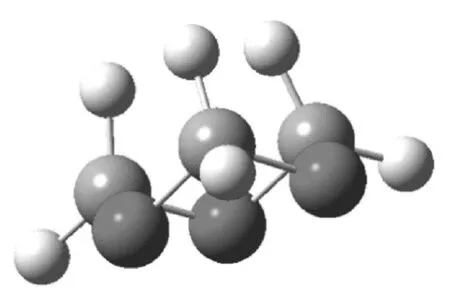
圖1 1,3,5-三氧六環的椅式構象
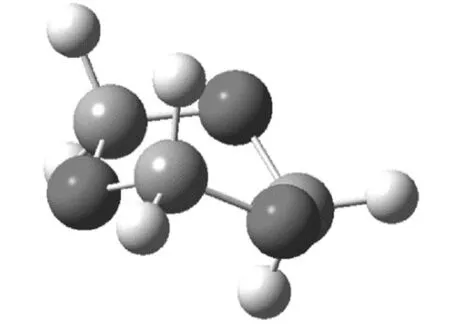
圖2 1,3,5-三氧六環的船式構象
圖3 正己烷構象
圖4 聚甲醛結構單元構象圖
各種六元環開環前后能量變化列于表2中,從表2可以看出,對于環己烷,其開環前的自由能更低,加上開環聚合過程是熵減少的過程,因此環己烷不可能發生開環聚合反應。四氫吡喃、1,4-二氧六環和1,3-二氧六環,有相似的情況,開環前后內能變化不大,考慮到聚合物熵的減少,自由能變化接近于零,小于理論計算的誤差,因而判斷為基本不能聚合,即使開環,也不能得到大分子量的聚合物。而1,3,5-三氧六環的自由能變化達到了-94 kJ·mol-1,因而是比較合適的開環聚合單體。
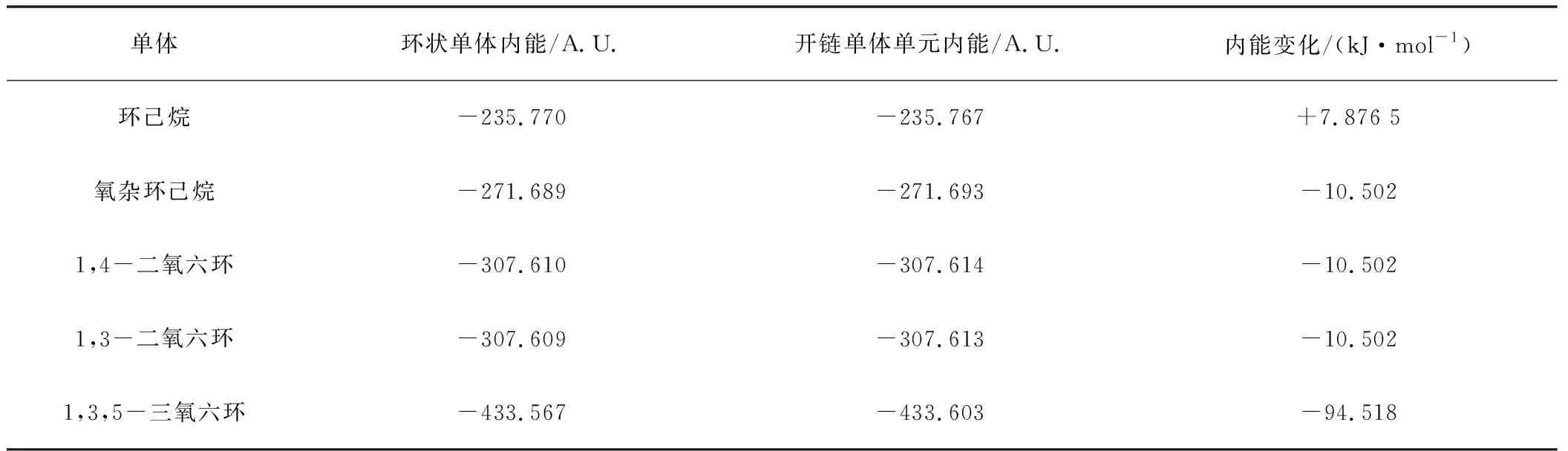
表2 六元環單體開環動力學數據
三氧六環開環的能量的變化,有幾個方面的原因,從表1可知,一方面,船式構象與椅式構象相比,不論是C-O-C鍵角還是O-C-C的鍵角,都有所增加,結果是氧原子之間的距離變大,這減少了電負性較大的氧原子之間的斥力,另外因為,直立氫原子之間的距離也變大,這兩方面的原因都使船式構象的能量有所降低,計算表明船式構象比椅式構象的能量要低86.6 kJ·mol-1。開鏈結構的聚甲醛,不是鋸齒狀結構,而是螺旋形結構,與船式構象的單體相比,一方面,盡管由于螺旋結構,氧原子距離沒有明顯比船式構象大,但不論是C-O-C鍵角還是O-C-C的鍵角,都有進一步增加, 這使得角張力大大減少,同時,直立氫原子的距離也大大地大于椅式構象中的氫原子的距離。盡管比船式構象中氫原子之間的距離稍小,但由于氫原子本身比較小,這個距離上其之間的斥力已經很小。同時,有斥力的直立氫原子的數量由六對減少為4對,使得螺旋形聚合物的能量較低。而鋸齒狀的聚合物結構中,一方面,C-O-C鍵角和O-C-C的鍵角,都比螺旋形結構的角度小,這使得角張力增大,同時,氧原子的距離也縮小了。另一方面,雖然氫原子之間的距離稍有增加,但是相斥的氫原子數量與螺旋形構想相比,則由2對變為6對。這兩方面的原因都導致鋸齒狀構象的能量高于螺旋形構象。
3 結論
與環己烷相比,四氫吡喃、1,3-二氧六環、1,4-二氧六環四種結構相似,其分子的最低能量構象為椅式,而1,3,5-三氧六環分子的最低能量構象為船式構象。前面四種單體開環后的鏈式結構的最低能量構象為鋸齒狀構象,而聚甲醛的最低能量構象為螺旋形。在聚甲醛螺旋形構象中,由于C-O-C鍵角和O-C-C的鍵角增加導致角張力減少,同時氧原子距離較大,另外,相斥的直立鍵上的氫原子只有兩對(其他結構中都是六對),這些因素都導致螺旋形構象的能量降低。計算表明,環己烷開環反應的自由能增加,因此環己烷通常不能發生開環聚合,四氫吡喃、1,3-二氧六環和1,4-二氧六環三種單體開環反應的自由能幾乎為零,所以一般情況下,也不能發生開環聚合,而1,3,5-三氧六環開環聚合的自由能變化為-94.5 kJ·mol-1,可以在催化劑作用下,順利進行聚合反應。
