基于棒狀核-殼結(jié)構(gòu)的磁性沸石咪唑酯骨架復(fù)合材料的磁固相萃取-氣相色譜-質(zhì)譜法檢測(cè)茶飲料中16 種多環(huán)芳烴
2024-10-17 00:00:00聶文丁帥思婕胡珂崔曉壘王靜李東東李騰飛
分析化學(xué) 2024年9期


摘要 制備了具有棒狀核-殼結(jié)構(gòu)的磁性沸石咪唑酯骨架復(fù)合材料(MHNTs@PDA@ZIF-8),并將其作為磁固相萃取(MSPE)的吸附劑,結(jié)合氣相色譜-質(zhì)譜聯(lián)用(GC-MS)技術(shù),建立了一種測(cè)定茶飲料中16種多環(huán)芳烴(PAHs)的方法。采用掃描電子顯微鏡(SEM)、傅里葉紅外光譜(FT-IR)、X射線衍射儀(XRD)、振動(dòng)樣品磁強(qiáng)計(jì)(VSM)和氮?dú)馕?脫附實(shí)驗(yàn)對(duì)此復(fù)合材料進(jìn)行了表征,結(jié)果表明,制備的MHNTs@PDA@ZIF-8吸附劑具有較強(qiáng)的磁性和較大的比表面積。考察了吸附劑用量、萃取時(shí)間和解吸時(shí)間等因素對(duì)萃取效率的影響,得到最佳萃取條件為:以10 mg MHNTs@PDA@ZIF-8作為吸附劑,渦旋萃取90 s, 1 mL正己烷超聲解吸60 s。本方法對(duì)16種PAHs在5~500 μg/L范圍內(nèi)具有良好的線性關(guān)系,相關(guān)系數(shù)r2≥0.995,檢出限(S/N=3)為0.1~0.8 μg/L, 定量限(S/N=10)為0.3~2.6 μg/L, 加標(biāo)回收率為60.9%~114.7%,相對(duì)標(biāo)準(zhǔn)偏差(RSD)為0.2%~9.2%。本方法操作簡(jiǎn)單、萃取時(shí)間短、靈敏度高、樣品和有機(jī)溶劑消耗少,適用于茶飲料中16 種PAHs的同時(shí)檢測(cè)。
關(guān)鍵詞 沸石咪唑酯骨架;埃洛石納米管;多環(huán)芳烴;磁固相萃取;茶飲料
多環(huán)芳烴(Polycyclic aromatic hydrocarbons, PAHs)是一類由兩個(gè)或兩個(gè)以上苯環(huán)組成的有機(jī)化合物,具有潛在的致癌、致畸和致突變效應(yīng)[1-2]。PAHs通常由有機(jī)物的不完全燃燒或高溫?zé)峤庑纬桑哂谐志眯浴⒁走w移,且能夠通過(guò)食物鏈富集于人體內(nèi),威脅人類健康,美國(guó)環(huán)保署(EPA)將16種PAHs列為優(yōu)先控制污染物[3-4]。茶葉在種植和干燥過(guò)程中可能受PAHs 污染,殘留的污染物進(jìn)一步轉(zhuǎn)移到茶飲料中,可能引起食品安全問(wèn)題[5]。因此,開發(fā)一種快速、準(zhǔn)確檢測(cè)茶飲料中PAHs的分析方法十分必要。
目前, PAHs的分析方法多采用氣相色譜-質(zhì)譜聯(lián)用法(GC-MS)[6]。由于PAHs具有強(qiáng)疏水性且在樣品中處于痕量水平,難以直接定量分析,需要對(duì)樣品進(jìn)行預(yù)處理,以有效富集目標(biāo)物、降低基質(zhì)干擾、提高檢測(cè)靈敏度和準(zhǔn)確度[7-8]。PAHs的樣品預(yù)處理方法有固相萃取(SPE)[9]、固相微萃取(SPME)[10]、磁性固相萃取(MSPE)[11]和分散固相萃取(DSPE)[12]等。其中, MSPE以功能化的磁性材料為吸附劑,通過(guò)引入外部磁場(chǎng)實(shí)現(xiàn)快速分離[13],具有吸附容量大、萃取效率高以及操作簡(jiǎn)便等優(yōu)點(diǎn)[14],并且磁性吸附劑易回收,可重復(fù)使用。高效的磁性吸附劑是MSPE 技術(shù)的核心[15],氧化石墨烯(GO)[16]、多壁碳納米管(MWCNTs)[17]、石墨氮化碳(g-C3N4)[18]以及金屬有機(jī)骨架(MOFs)[19]等納米材料已經(jīng)成功用于MSPE。MOFs 是由金屬離子/團(tuán)簇和有機(jī)配體配位而成的有機(jī)-無(wú)機(jī)雜化材料,具有比表面積大、穩(wěn)定性好、孔隙率高和易修飾等優(yōu)點(diǎn)[20-21]。ZIF-8是沸石咪唑酯骨架(Zeolitic imidazolate frameworks, ZIFs)材料的典型代表[22],由鋅離子和2-甲基咪唑自組裝而成,具有疏水性和π 電子共軛體系。選擇合適的載體材料對(duì)ZIF-8進(jìn)行固定化,可望實(shí)現(xiàn)多種材料的優(yōu)勢(shì)疊加[23]。四氧化三鐵(Fe3O4)納米顆粒具有超順磁性,通常作為磁性載體進(jìn)行功能化修飾。埃洛石納米管(Halloysite nanotubes, HNTs)是一種天然的鋁硅酸鹽管狀粘土材料,具有穩(wěn)定性高、比表面積大、分散性好且廉價(jià)易得等優(yōu)點(diǎn)[24-25]。
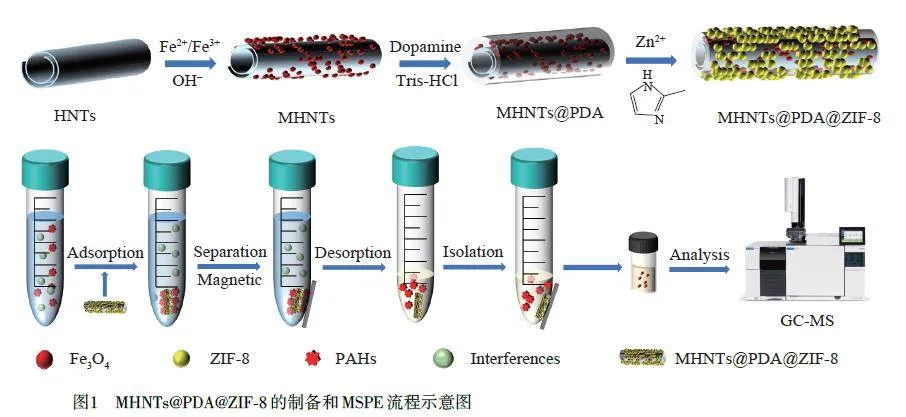
本研究以HNTs 為基底材料,通過(guò)化學(xué)共沉淀法在HNTs 上沉積Fe3O4,以減少Fe3O4 納米顆粒團(tuán)聚,增強(qiáng)磁性材料的分散性。在弱堿性溶液中,多巴胺(DA)通過(guò)氧化自聚合成聚多巴胺(PDA)將磁性埃洛石納米管(MHNTs)包裹,然后鋅離子通過(guò)螯合作用被錨定在MHNTs@PDA 表面,最后將其置于含二甲基咪唑的體系中,誘導(dǎo)ZIF-8 在MHNTs@PDA 表面成核和生長(zhǎng),合成了具有棒狀核-殼結(jié)構(gòu)的MHNTs@PDA@ZIF-8復(fù)合材料。此復(fù)合材料的ZIF-8具有π 電子共軛體系,可與芳香族化合物PAHs形成較強(qiáng)的π-π 相互作用;MHNTs結(jié)合了HNTs的高吸附容量和Fe3O4 快速分離的優(yōu)點(diǎn),同時(shí)與ZIF-8具有協(xié)同效應(yīng)。將此復(fù)合材料作為MSPE 吸附劑,結(jié)合GC-MS 技術(shù)建立了一種快速檢測(cè)茶飲料中16 種PAHs的MSPE/GC-MS方法(圖1)。
1 實(shí)驗(yàn)部分
1.1 儀器與試劑
8890-5977B氣相色譜-質(zhì)譜聯(lián)用儀(美國(guó)Agilent公司);D8-ADVANCE X射線衍射儀(德國(guó)布魯克公司);Lake Shore 7404振動(dòng)樣品磁強(qiáng)計(jì)(美國(guó)Lakeshore公司);Nicolet 6700傅里葉紅外光譜儀(美國(guó)尼高力公司);Hitachi S-4800 場(chǎng)發(fā)射掃描電子顯微鏡(日本日立公司);ASAP 2460 全自動(dòng)比表面積及孔徑分析儀(美國(guó)麥克公司)。
16種PAHs標(biāo)準(zhǔn)溶液(1000 mg/L, 溶劑為正己烷-丙酮(1∶1,V/V))購(gòu)于上海安譜實(shí)驗(yàn)科技股份有限公司,包括:萘(Nap)、苊烯(Acpy)、苊(Acen)、芴(Flu)、菲(Phen)、蒽(Ant)、熒蒽(Fluo)、芘(Pyr)、苯并[a]蒽(B[a]A)、?(Chr)、苯并[b]熒蒽(B[b]F)、苯并[k]熒蒽(B[k]F)、苯并[a]芘(B[a]P)、茚并[1,2,3-cd]芘(I[1,2,3-cd]P)、二苯并[a,h]蒽(D[a,h]A)和苯并[g,h,i]芘(B[g,h,i]P)。
正己烷、二氯甲烷、苯、甲苯、乙酸乙酯和甲醇(色譜純,上海安譜實(shí)驗(yàn)科技股份有限公司);HNTs(分析純,廣州潤(rùn)沃材料科技有限公司);鹽酸多巴胺、FeCl3·6H2O和2-甲基咪唑和三羥甲基氨基甲烷(Tris)(分析純,上海阿拉丁試劑有限公司);FeCl2·4H2O(分析純,上海麥克林生化科技有限公司); (CH3COO)2Zn·2H2O(分析純,天津巴斯夫化工有限公司);無(wú)水乙醇(分析純,天津永大化學(xué)試劑有限公司);氨水(25%,分析純,天津大茂化學(xué)試劑有限公司);HCl(36%)、NaCl 和NaOH(分析純,國(guó)藥集團(tuán)化學(xué)試劑有限公司)。實(shí)驗(yàn)用水為超純水(18.2 MΩ·cm)。
1.2 MHNTs@PDA@ZIF-8 復(fù)合材料的制備
1.2.1 MHNTs 的制備
參考文獻(xiàn)[26]的方法制備MHNTs。將2.5 g HNTs 加入到150 mL 水中,超聲分散10 min;加入5.41 g的FeCl3·6H2O和4.00 g的FeCl2·4H2O,在60 ℃ 下攪拌30 min;緩慢加入50 mL 25% 氨水,70 ℃ 下攪拌4 h;產(chǎn)物經(jīng)磁分離后,用水和無(wú)水乙醇洗滌至中性, 60 ℃ 下真空干燥,得到MHNTs。
1.2.2 MHNTs@PDA 的制備
將0.4 g MHNTs均勻分散在100 mL Tris-HCl緩沖溶液(10 mmol/L, pH=8.5)中,加入0.2 g鹽酸多巴胺,室溫下攪拌反應(yīng)6 h;產(chǎn)物經(jīng)磁分離后,依次用水和無(wú)水乙醇洗滌3次,在60 ℃ 下真空干燥,即得到MHNTs@PDA。
1.2.3 MHNTs@PDA@ZIF-8 的制備
將1.1 g (CH3COO)2Zn·2H2O溶于70 mL甲醇中,加入0.4 g MHNTs@PDA,超聲分散10 min;向上述溶液中加入70 mL含3.3 g 2-甲基咪唑的甲醇溶液,室溫下攪拌3 h;產(chǎn)物經(jīng)磁分離后,用甲醇洗滌3次,60 ℃ 下真空干燥,得到MHNTs@PDA@ZIF-8。
1.3 MSPE 程序
準(zhǔn)確移取1 mL 樣品,用超純水稀釋至10 mL, 加入0.2 g NaCl 調(diào)節(jié)離子強(qiáng)度。向此溶液中加入10 mg MHNTs@PDA@ZIF-8吸附劑,將混合物渦旋90 s以達(dá)到吸附平衡;采用外部磁鐵分離吸附劑,棄去溶液;加入1 mL正己烷超聲解吸60 s,磁分離,溶液過(guò)0.22 μm有機(jī)濾膜后,進(jìn)行GC-MS分析。
1.4 GC-MS 條件
1.4.1 GC 條件
色譜柱:HP-5 MS 毛細(xì)管柱(30 m×0.25 mm×0.25 μm, 美國(guó)Agilent 公司);載氣:高純氦氣(純度≥ 99.999 %);流速:1.2 mL/min;進(jìn)樣量:1 μL;進(jìn)樣口溫度:280 ℃, 采用不分流進(jìn)樣模式。升溫程序:80 ℃ 保持2 min;以10 ℃/min速率升至280 ℃, 保持3 min;以10 ℃/min速率升溫至300 ℃,保持2 min;以20 ℃/min速率升溫至320 ℃, 保持3 min。
1.4.2 MS 條件
離子源:電子轟擊源(EI);電離電壓:70 eV;四極桿溫度:150 ℃;離子源溫度:230 ℃;傳輸線溫度:280 ℃;溶劑延遲:6 min;選擇離子監(jiān)測(cè)(SIM)模式采集。
2 結(jié)果與討論
2.1 合成材料表征
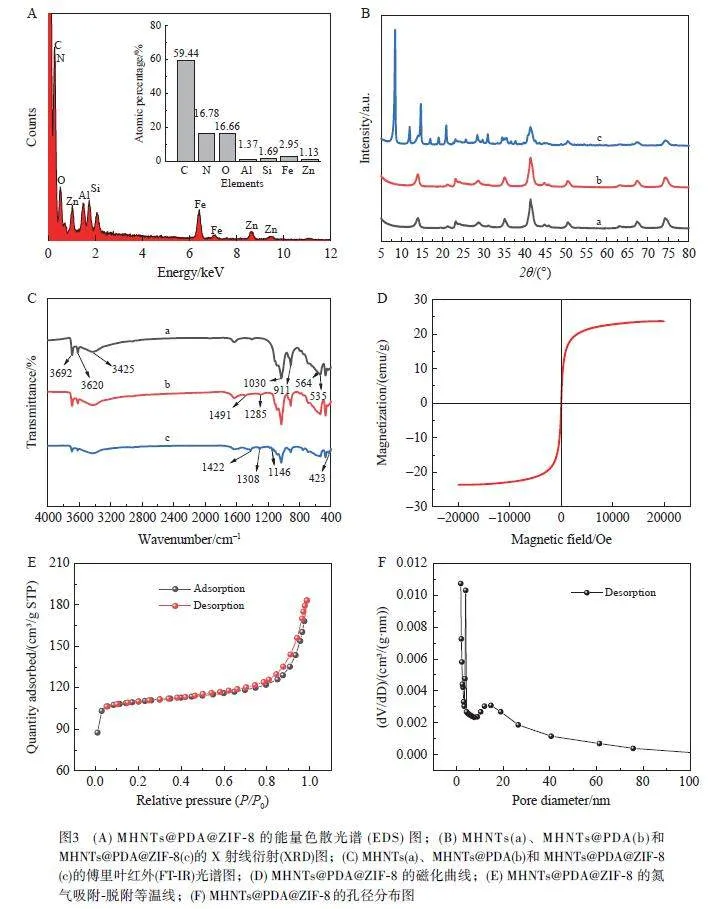
采用掃描電子顯微鏡(SEM)對(duì)MHNTs、MHNTs@PDA和MHNTs@PDA@ZIF-8材料進(jìn)行形貌表征。如圖2A 所示, Fe3O4 納米顆粒成功負(fù)載在HNTs 表面,部分顆粒由于高磁化強(qiáng)度而團(tuán)聚;由圖2B 可見,引入PDA 后不影響MHNTs 的結(jié)構(gòu),可以為后續(xù)ZIF-8 的負(fù)載提供支撐;由圖2C 可見,外壁生長(zhǎng)了ZIF- 8 納米顆粒, 呈典型的十二面體結(jié)構(gòu)。如圖3A 所示, 在能量色散光譜( EDS) 圖中存在C、N、O、Al、Si、Fe和Zn 7種元素,與MHNTs@PDA@ZIF-8復(fù)合材料的元素組成一致。
采用X射線衍射(XRD)對(duì)MHNTs、MHNTs@PDA和MHNTs@PDA@ZIF-8的晶體結(jié)構(gòu)進(jìn)行表征。如圖3B所示,MHNTs(曲線a)在2θ=21.3°、35.1°、41.4°、50.5°、63.2°、67.4° 和74.4° 處的衍射峰對(duì)應(yīng)于Fe3O4 的特征峰。與MHNTs相比,MHNTs@PDA(曲線b)的衍射峰未改變,并且未出現(xiàn)其它雜峰,表明經(jīng)PDA 改性后,產(chǎn)物的晶型并未改變。在MHNTs@PDA@ZIF-8(曲線c)中, 2θ=8.5°、12.0°、14.8°、17.0°、19.0° 和25.7° 處的衍射峰對(duì)應(yīng)于ZIF-8的特征峰,表明ZIF-8成功制備。
MHNTs、MHNTs@PDA和MHNTs@PDA@ZIF-8的傅里葉紅外(FT-IR)光譜如圖3C所示,在MHNTs的FT-IR譜圖(曲線a)中, 3692和3620 cm–1 處的特征峰歸屬于HNTs的Si—OH的伸縮振動(dòng),1030 cm–1 處的特征峰歸屬于Si—O的伸縮振動(dòng);535和911 cm–1 處的特征峰分別歸屬于Al—O—Si的變形振動(dòng)和內(nèi)部Al—OH的振動(dòng),3425和564 cm–1 處的特征峰分別歸屬于O—H的伸縮振動(dòng)和Fe—O的伸縮振動(dòng),表明Fe3O4 成功負(fù)載在HNTs上[26];在MHNTs@PDA的FT-IR譜圖(曲線b)中,1491 cm–1 處中新增的特征峰歸屬于芳香環(huán)的伸縮振動(dòng),1285 cm–1 處的特征峰代表C—N 的伸縮振動(dòng)峰,表明PDA 成功涂覆在MHNTs上;在MHNTs@PDA@ZIF-8的FT-IR譜圖(曲線c)中,1422 cm–1 處出現(xiàn)的特征峰歸屬于咪唑環(huán)的伸縮振動(dòng),1308 和1146 cm–1 處的特征峰為咪唑環(huán)的彎曲振動(dòng)峰, 423 cm–1 處的特征峰為Zn—N 伸縮振動(dòng)峰。FT-IR光譜結(jié)果表明MHNTs@PDA@ZIF-8被成功制備。
對(duì)MHNTs@PDA@ZIF-8進(jìn)行磁性能分析,結(jié)果如圖3D 所示,MHNTs@PDA@ZIF-8飽和磁化強(qiáng)度為23.70 emu/g,表明此材料具有較強(qiáng)的磁性,可以實(shí)現(xiàn)快速分離。圖3E和3F分別為MHNTs@PDA@ZIF-8的氮?dú)馕?脫附等溫線和孔徑分布圖,圖中等溫線屬于Ⅳ型等溫線, BET比表面積為347.61 m2/g,平均孔徑為3.26 nm, 孔體積為0.283 cm3/g,表明此材料存在介孔結(jié)構(gòu),并且具有較大的比表面積,可以提供更多的吸附位點(diǎn)。
2.2 MSPE 條件的優(yōu)化
考察了MSPE參數(shù)包括MHNTs@PDA@ZIF-8吸附劑用量、萃取時(shí)間、離子強(qiáng)度、解吸時(shí)間、解吸溶劑類型和解吸溶劑體積對(duì)萃取效率的影響。采用10 mL加標(biāo)水樣(100 μg/L)進(jìn)行優(yōu)化實(shí)驗(yàn),每組實(shí)驗(yàn)平行測(cè)定3次。
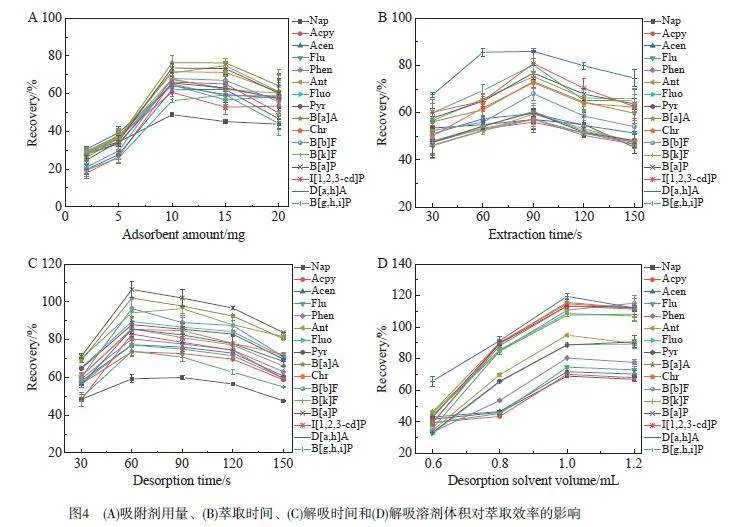
首先考察了MHNTs@PDA@ZIF-8 吸附劑用量(2~20 mg)對(duì)萃取效率的影響。如圖4A 所示,當(dāng)吸附劑用量從2 mg增至10 mg 時(shí), PAHs的回收率明顯提升,進(jìn)一步增加吸附劑用量對(duì)萃取效率沒有顯著影響,表明10 mg的吸附劑足以完全吸附PAHs。因此,選擇MSPE的最佳吸附劑用量為10 mg。
萃取時(shí)間和解吸時(shí)間(30~150 s)對(duì)萃取效率的影響如圖4B所示,當(dāng)萃取時(shí)間從30 s延長(zhǎng)至90 s時(shí),PAHs的回收率緩慢增加,在90 s時(shí)達(dá)到最大值。隨著萃取時(shí)間進(jìn)一步延長(zhǎng),可能由于反擴(kuò)散作用而導(dǎo)致回收率下降。因此,選擇最佳萃取時(shí)間為90 s。當(dāng)解吸時(shí)間由30 s延長(zhǎng)至60 s時(shí)(圖4C), PAHs的回收率逐漸增大,并且在60 s時(shí)達(dá)到最大。因此,最佳解吸時(shí)間選擇60 s。
通過(guò)添加0~15%(m/V)的NaCl考察離子強(qiáng)度對(duì)萃取效率的影響。當(dāng)NaCl濃度增加到2%(m/V)時(shí),PAHs的回收率最高(見電子版文后支持信息圖S1),推測(cè)可能是NaCl的加入降低了PAHs在樣品溶液中的溶解度,從而增強(qiáng)了吸附劑對(duì)PAHs的吸附能力。過(guò)量的NaCl會(huì)增加溶液的黏度,并占據(jù)吸附劑的活性位點(diǎn),不利于PAHs吸附。因此,本研究選擇向樣品溶液中添加2%(m/V)NaCl溶液。
考察了解吸溶劑類型和體積對(duì)萃取效率的影響。選擇5種有機(jī)溶劑(正己烷、二氯甲烷、甲苯、乙酸乙酯和苯)評(píng)價(jià)其萃取效率。以正己烷為解吸溶劑時(shí), PAHs的萃取效率最高(見電子版文后支持信息圖S1)。解吸溶劑體積直接影響目標(biāo)物的回收率,解吸溶劑過(guò)少可能會(huì)因?yàn)橄疵摬煌耆鴮?dǎo)致分析物回收率低,而過(guò)量的解吸溶劑會(huì)降低方法的靈敏度。如圖4D 所示,當(dāng)解吸溶劑體積從0.6 mL 增至1.0 mL時(shí),PAHs的回收率逐漸增大,在1.0 mL時(shí)達(dá)到最佳值。因此,本研究選擇1.0 mL正己烷進(jìn)行解吸。
2.3 吸附性能考察
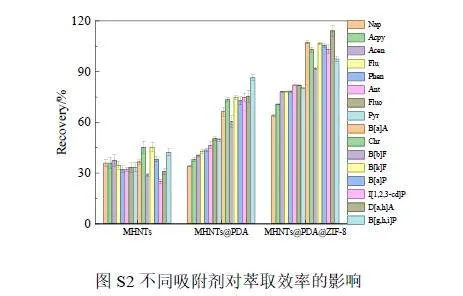
比較了MHNTs、MHNTs@PDA和MHNTs@PDA@ZIF-8這3種吸附劑對(duì)目標(biāo)分析物的吸附性能,結(jié)果表明, MHNTs@PDA@ZIF-8 對(duì)PAHs 的萃取效果最好(見電子版文后支持信息圖S2)。這可歸因于MHNTs@PDA@ZIF-8具有π 電子共軛體系、疏水性以及大的比表面積,可通過(guò)π-π 堆積和疏水作用等增強(qiáng)其與PAHs的相互作用,從而提高對(duì)PAHs的萃取效率。
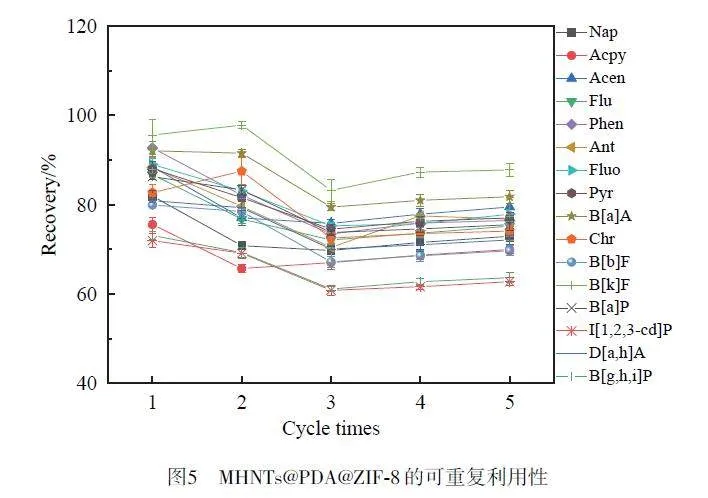
在最佳條件下,評(píng)估了MHNTs@PDA@ZIF-8的可重復(fù)利用性。每次吸附-解吸循環(huán)后,使用2 mL正己烷將MHNTs@PDA@ZIF-8 重復(fù)洗脫2 次,以消除殘留物的干擾。如圖5 所示,重復(fù)使用5 次后,16 種PAHs的回收率均仍能達(dá)到60% 以上,表明制備的MHNTs@PDA@ZIF-8穩(wěn)定性好,至少可重復(fù)使用5次。
2.4 基質(zhì)效應(yīng)評(píng)價(jià)
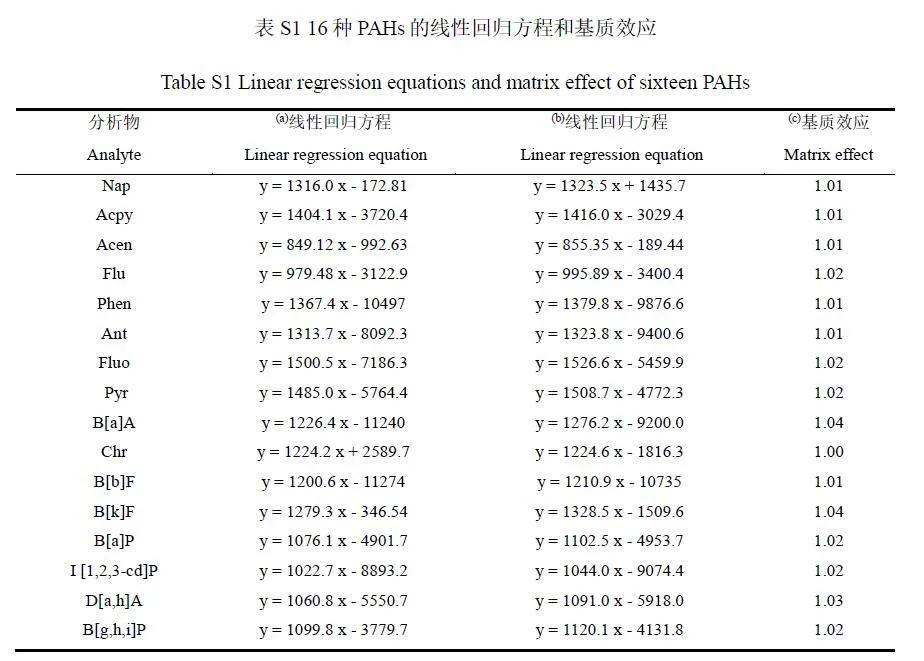
分別用溶劑和基質(zhì)空白液配制系列標(biāo)準(zhǔn)曲線,通過(guò)基質(zhì)匹配標(biāo)準(zhǔn)曲線和溶劑標(biāo)準(zhǔn)曲線的斜率比值評(píng)價(jià)基質(zhì)效應(yīng)強(qiáng)弱。當(dāng)斜率比lt;1 時(shí),為基質(zhì)抑制效應(yīng);當(dāng)斜率比gt;1 時(shí),為基質(zhì)增強(qiáng)效應(yīng);當(dāng)0.8≤斜率比≤1.2時(shí),通常認(rèn)為是弱基質(zhì)效應(yīng),可以忽略不計(jì)[27]。結(jié)果表明,16種PAHs的基質(zhì)效應(yīng)在1.00~1.04范圍內(nèi)(見電子版文后支持信息表S1),基質(zhì)效應(yīng)可以忽略,故采用溶劑配制標(biāo)準(zhǔn)曲線。
2.5 方法學(xué)考察
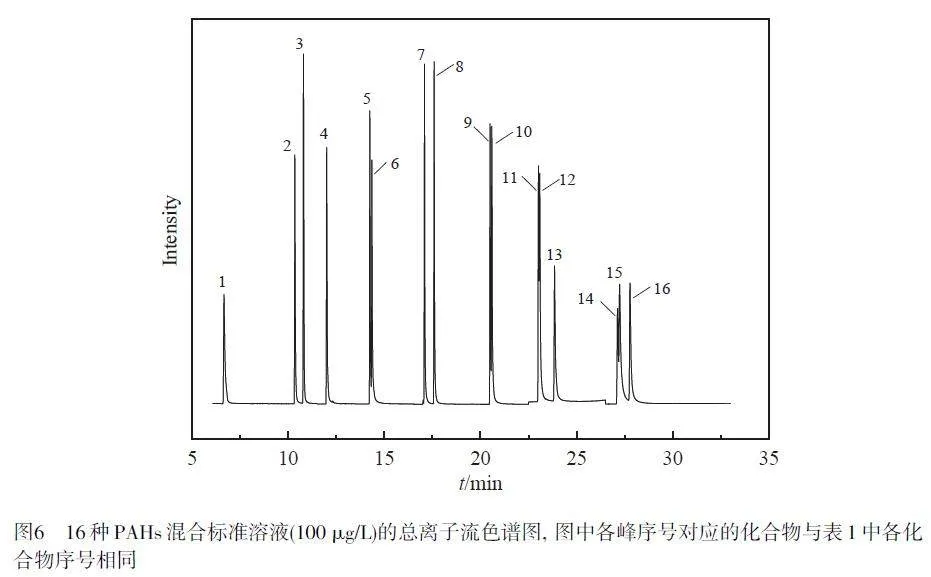
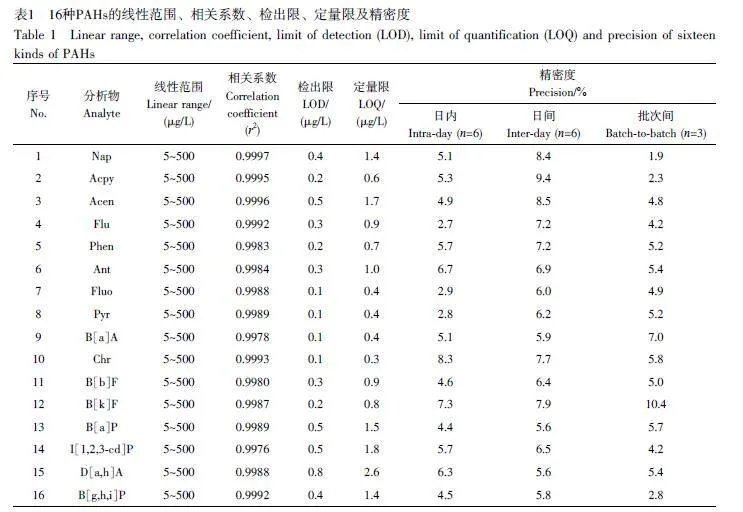
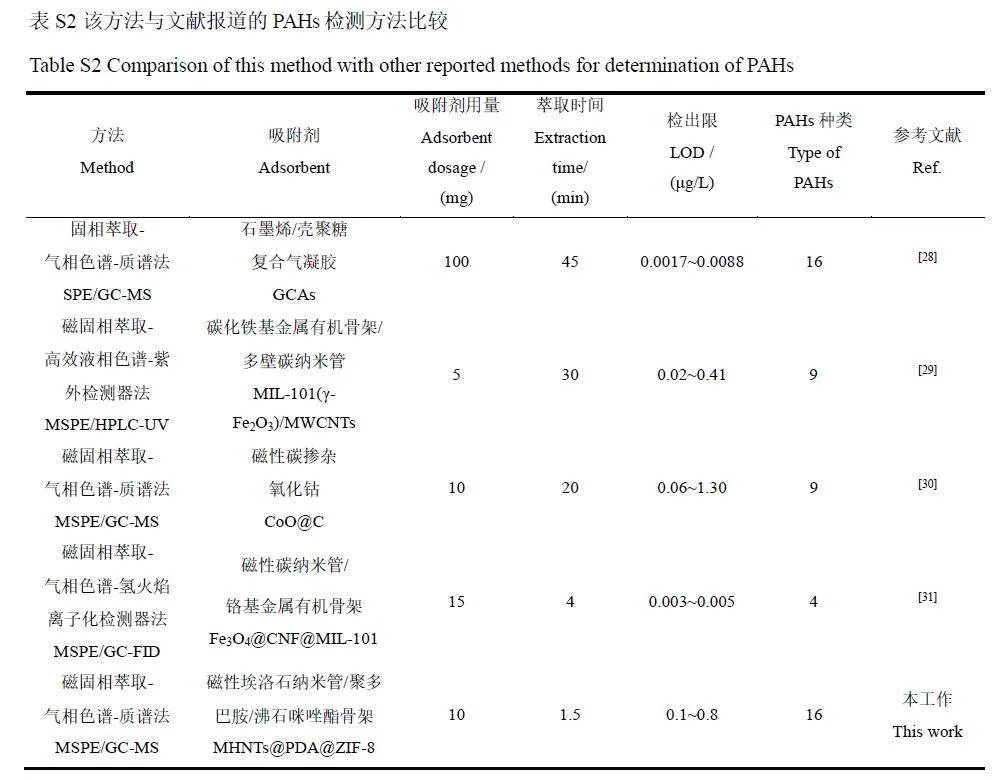
配制16 種PAHs 的系列標(biāo)準(zhǔn)溶液,在最佳條件下采用GC-MS 進(jìn)行分析,以PAHs 質(zhì)量濃度為橫坐標(biāo)、對(duì)應(yīng)的峰面積為縱坐標(biāo),繪制標(biāo)準(zhǔn)曲線,總離子流色譜圖(TIC)見圖6。在5~500 μg/L范圍內(nèi),16種PAHs 線性關(guān)系良好,相關(guān)系數(shù)(r2)≥0.995,方法檢出限(S/N=3)為0.1~0.8 μg/L, 定量限(S/N=10)為0.3~2.6 μg/L(表1)。采用加標(biāo)茶飲料(100 μg/L)評(píng)估日內(nèi)、日間和3批次材料之間的精密度,16種PAHs的日內(nèi)精密度為2.7%~8.3%,日間精密度為5.6%~9.4%,3批次材料之間的精密度為1.9%~10.4%,這表明本方法具有良好的穩(wěn)定性和重復(fù)性。與文獻(xiàn)報(bào)道的PAHs檢測(cè)方法相比(電子版文后支持信息表S2),本方法滿足16種PAHs同時(shí)測(cè)定的要求,具有萃取時(shí)間短和吸附劑用量少等優(yōu)點(diǎn),是一種很有潛力的富集和檢測(cè)食品中PAHs的方法。
2.6 實(shí)際樣品分析
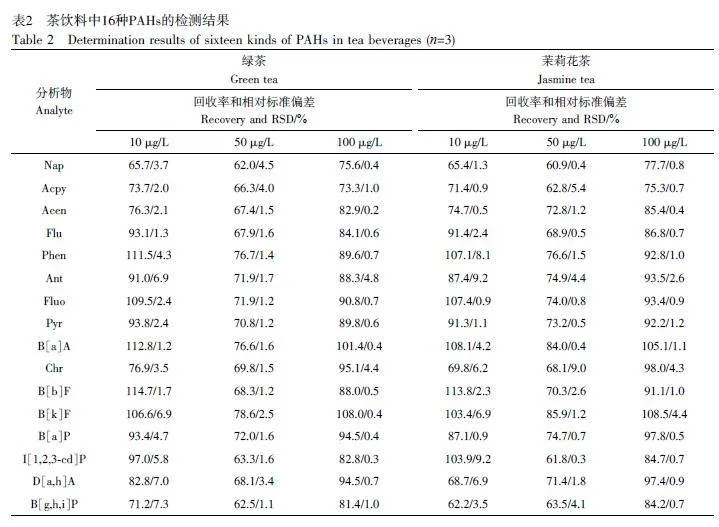
選擇市售的茉莉花茶飲料和綠茶飲料作為實(shí)際樣品進(jìn)行分析,均未檢測(cè)出PAHs。向樣品中分別添加3個(gè)濃度水平(10、50和100 μg/L)的PAHs標(biāo)準(zhǔn)溶液進(jìn)行加標(biāo)回收實(shí)驗(yàn),每個(gè)濃度平行測(cè)定3次。結(jié)果見表2,平均加標(biāo)回收率為60.9%~114.7%,相對(duì)標(biāo)準(zhǔn)偏差(RSD)為0.2%~9.2%。上述結(jié)果表明,本方法在檢測(cè)茶飲料中PAHs方面具有實(shí)際應(yīng)用潛能。
3 結(jié)論
采用簡(jiǎn)單且環(huán)保的方法合成了一種比表面積大、結(jié)構(gòu)穩(wěn)定的MHNTs@PDA@ZIF-8 復(fù)合材料,此材料對(duì)PAHs具有良好的吸附效果。將MHNTs@PDA@ZIF-8用于MSPE過(guò)程,結(jié)合GC-MS技術(shù),建立了茶飲料中16 種PAHs 的MSPE/GC-MS 分析方法,此方法操作簡(jiǎn)便、萃取時(shí)間短、溶劑用量少、準(zhǔn)確度高,為食品中痕量PAHs的檢測(cè)提供了技術(shù)參考。
References
[1] BARATHI S, J G, RATHINASAMY G, SABAPATHI N, ARULJOTHI K N, LEE J, KANDASAMY S. Chemosphere, 2023,337: 139396.
[2] SUN K, SONG Y, HE F, JING M, TANG J, LIU R. Sci. Total Environ. , 2021, 773: 145403.
[3] MALLAH M A, CHANGXING L, MALLAH M A, NOREEN S, LIU Y, SAEED M, XI H, AHMED B, FENG F, MIRJAT A A, WANG W, JABAR A, NAVEED M, LI J H, ZHANG Q. Chemosphere, 2022, 296: 133948.
[4] CHEN B H, INBARAJ B S, HSU K C. J. Food Drug Anal. , 2022, 30(4): 494-522.
[5] ZHOU D B, SHENG X, HAN F, HU Y Y, DING L, LV Y L, SONG W, ZHENG P. J. Chromatogr. A, 2018, 1578: 53-60.
[6] FAN J, YAO X, YAN Z, LI Y, LIU J, CAI Y. Microchem. J. , 2023, 195: 109444.
[7] SHI Y, WU H, WANG C, GUO X, DU J, DU L. Food Chem. , 2016, 199: 75-80.
[8] DENG W, HUANG A, ZHENG Q, YU L, LI X, HU H, XIAO Y. Food Chem. , 2021, 352: 129331.
[9] SHI L, ZHENG L, LIU R, CHANG M, HUANG J, JIN Q, WANG X. Food Chem. , 2019, 275: 206-213.
[10] YU C, ZHANG J, LUO X, ZHANG J. Microchem. J. , 2023, 187: 108388.
[11] TONG Y, LI S, WU Y, GUO J, ZHOU B, ZHOU Q, JIANG L, NIU J, ZHANG Y, LIU H, YUAN S, HUANG S, ZHAN Y.Chemosphere, 2022, 296: 134009.
[12] LI Y, YAN Z, FAN J, YAO X, CAI Y. Talanta, 2023, 265: 124916.
[13] HASSAN F W M, RAOOV M, KAMARUZAMAN S, MOHAMED A H, IBRAHIM W N W, HANAPI N S M, ZAIN N N M,YAHAYA N, CHEN D D Y. J. Food Compos. Anal. , 2021, 102: 104057.
[14] LI G, WEN A, LIU J, WU D, WU Y. Food Chem. , 2021, 337: 127974.
[15] HUANG Si-Min, HU Yu-Fei, CHEN Yan-Long, LI Gong-Ke, XIA Ling. Chin. J. Anal. Chem. , 2020, 48(10): 1392-1399.
黃斯敏, 胡玉斐, 陳彥龍, 李攻科, 夏凌. 分析化學(xué), 2020, 48(10): 1392-1399.
[16] ZHU L, XU H. J. Sep. Sci. , 2014, 37(18): 2591-2598.
[17] MOAZZEN M, AHMADKHANIHA R, GORJI M E, YUNESIAN M, RASTKARI N. Talanta, 2013, 115: 957-965.
[18] WANG M, CUI S, YANG X, BI W. Talanta, 2015, 132: 922-928.
[19] ZHOU Q, LEI M, LI J, LIU Y, ZHAO K, ZHAO D. Microchim. Acta, 2017, 184(4): 1029-1036.
[20] LIU G, ZHANG X, LU M, TIAN M, LIU Y, WANG J, LI L, LI T, CHEN G, XU D. Food Chem. , 2022, 393: 133337.
[21] SHI W, LI W, NGUYEN W, CHEN W, WANG J, CHEN M. Mater. Today Adv. , 2022, 15: 100273.
[22] ZHANG X, YUAN N, XU S, LI Y, WANG Q. J. Ind. Eng. Chem. , 2022, 111: 155-167.
[23] ZHU X, TONG J, ZHU L, PAN D. Int. J. Biol. Macromol. , 2022, 205: 473-482.
[24] LVOV Y, WANG W, ZHANG L, FAKHRULLIN R. Adv. Mater. , 2016, 28(6): 1227-1250.
[25] YUAN X, WEI Z, ZHANG Z, LIU H. J. Inorg. Organomet. Polym. Mater. , 2022, 32(8): 3030-3039.
[26] ZHOU W, WANG X, LIU Y, ZHANG W, DI X. Food Chem. , 2023, 404: 134432.
[27] ZHAO W H, SHI Y P. J. Chromatogr. A, 2022, 1670: 462968.
[28] GAO Y, QIN Y, XIONG F, ZHAO L. J. Sep. Sci. , 2020, 43(20): 3940-3948.
[29] YANG J, ZHANG X, WANG X, WANG H, ZHAO J, ZHOU Z, DU X, LU X. J. Chromatogr. A, 2022, 1681: 463459.
[30] HUANG P, KOU H, WANG X, ZHOU Z, DU X, LU X. Talanta, 2021, 227: 122149.
[31] BODAGHABADI F, AMIRI A, MIRZAEI M. Anal. Methods, 2023, 15(41): 5526-5534.
河北省重點(diǎn)研發(fā)計(jì)劃項(xiàng)目(No. 22325501D)資助。
