基于全基因組重測序分析大尾寒羊基因組變異特征和群體結構
2024-12-18 00:00:00梁慧麗解玉靜司博王桂英姜運良曹貴玲
畜牧獸醫學報 2024年11期
摘 要: 旨在了解大尾寒羊基因組遺傳變異特征和群體結構,可以為更好地保護和利用大尾寒羊提供指導。本研究對170只(66只公羊,104只母羊)大尾寒羊進行了全基因組重測序,利用GATK、Manta、Plink等軟件對大尾寒羊基因組遺傳變異、群體結構和連鎖不平衡等進行了分析,以期了解大尾寒羊基因組變異特征和群體結構。測序共獲得1 599.56 G高質量數據(平均9.409 G·只-1)。大尾寒羊群體中共發現50 276 079個SNPs和7 240 540個InDel,它們多分布于基因間和內含子區域。群體的基因組結構性變異(SV)最多的類型為染色體間的易位(CTX),平均每只羊有415.82個CTX,主要分布在基因間區域;發生拷貝數變異(CNV)最多的區域在外顯子,平均每只羊有175個。主成分分析顯示,大尾寒羊個體較分散,聚集不集中。結合親緣關系、系統發育樹和群體結構,將大尾寒羊分為6個家系,各家系含量差別較大,體型有差異。群體聚類分析中發現有些個體祖先成分較為單一。群體連鎖不平衡(LD)分析顯示LD衰減速度快,群體遺傳多樣性較高。馴化中受選擇的基因主要與脂質代謝和產熱有關。綜上,大尾寒羊群體包含6個家系,遺傳多樣性較豐富,保種效果良好,建議對小家系進行擴繁,大家系注意減少近交,確保家系結構平衡,同時注重大尾寒羊的開發和利用。
關鍵詞: 大尾寒羊;全基因組重測序;基因組變異;群體結構
中圖分類號: S826.2
文獻標志碼:A
文章編號:0366-6964(2024)11-4968-12
收稿日期:2024-03-26
基金項目:山東省自然科學基金(ZR2021MC130); 山東省農業良種工程(2019LZGC019)
作者簡介:梁慧麗(2000-),女,山東菏澤人,碩士生,主要從事動物遺傳育種研究,E-mail: 2210190109@stu.lcu.edu.cn
*通信作者:曹貴玲,主要從事動物遺傳育種與繁殖研究,E-mail: cglling@126.com
Analysis on Genomic Variation and Population Structure of Large-tailed Han Sheep Based
on Whole Genome Resequencing
LIANG" Huili1, XIE" Yujing1, SI" Bowen1, WANG" Guiying1, JIANG" Yunliang2, CAO" Guiling1,2*
(1.Agricultural Science and Engineering School, Liaocheng University, Liaocheng 252000," China;
2.Shandong Provincial Key Laboratory of Animal Biotechnology and Disease Control and Prevention,
Shandong Agriculture University, Taian 201718," China)
Abstract:" This study aimed to analyze the genomic variations and population structure of Large-tailed Han (LTH) sheep to provide guidance for scientific conservation and sustainable utilization. Whole-genomic sequencing data from 170 LTH sheep (66 rams, 104 ewes) was used to investigate the genomic variations, population structure and linkage disequilibrium by GATK, Manta, Plink and other softwares. The 1 599.56 G clean data was obtained and on average, 9.409 G clean data per sheep. In the LTH sheep population, 50 276 079 SNPs and 7 240 540 InDel were found, which were mostly distributed in intergenic and intronic region. The interchromosomal translocation (CTX) was the most common type of structural variation (SV) in the genome of LTH sheep, on average, 415.82 CTX per sheep, and also mainly distributed in the intergenic region. The copy number variation (CNV) was mostly distributed in exons, 175 CNV per sheep on average. The result of principal component analysis showed that individuals were relatively scattered and poorly clustered. According to the genetic relationship, phylogenetic tree and population structure, the 170 LTH sheep were clustered into 6 families, and there was a significant difference in family size and body size. The results of cluster analysis showed that ancestry component of some individuals was single. The linkage disequilibrium analysis showed that LTH sheep population had a relatively fast attenuation rate. The selected genes during domestication were mainly related with lipids metabolism and thermogenesis. In conclusion, LTH sheep population contains 6 families and has rich genetic diversity and remarkable conservation effect. It is suggested to expand the number of small families and to prevent inbreeding in the big families to keep a balanced population size and structure and focus on the utilization of LTH sheep.
Key words: Large-tailed Han sheep; whole genome resequencing; genomic variation; population structure
*Corresponding author:" CAO Guiling, E-mail: cglling@126.com
大尾寒羊(Large-tailed Han (LTH) sheep)屬肉脂兼用型地方綿羊品種,具有耐粗飼、抗病力強和肉脂性能好的特點。大尾寒羊是長脂尾品種,脂尾肥大,形似芭蕉扇,桃形尾尖上翻,緊貼于尾溝,下垂至飛節以下,有些個體尤其是公羊,尾接近或拖及地面[1]。綿羊的脂尾特征被認為是對惡劣環境的適應性反應,是綿羊在食物短缺時期的寶貴儲備[2]。近幾年,人們關注羊尾脂沉積的生理、生化和基因組調控,以揭示能量和脂質代謝在綿羊尾型形成中的潛在作用機制。有不少研究利用RNA-seq、基因組測序等技術分析不同尾型羊的尾脂轉錄組[3-4]和脂質成分[5]、腸道微生物差異[6]和基因組差異[7-8]等,發現了一些影響綿羊尾型的基因如PDGFD、POSTN、LGALS12、GGPS1和SETD4等。也有一些研究以大尾寒羊為研究材料對綿羊脂尾形成、尾脂沉積機制等進行了分析[9-13]。
在物質匱乏的年代,大尾寒羊的尾脂作為食物給人類提供了能量,但隨著生活水平的不斷提高,人們對尾脂的需求越來越少。另外,在現代化養殖模式下,大尾寒羊的長脂尾帶來了諸多不便,如不利于配種,加上飼料轉化率低等諸多原因,導致人們飼養大尾寒羊的意愿降低,大尾寒羊數量減少,目前僅存數百只。為了保護這一獨特的優良遺傳資源,我國已經建立了國家級和山東省省級大尾寒羊保種場進行活體保種,并進行了基因組和精液保存[14]。但目前大尾寒羊群體系譜記錄不完整,親緣關系模糊等問題對大尾寒羊保種造成了一定困難。如何更好地保存、開發和利用大尾寒羊是目前函待解決的問題。全基因組重測序基于高通量測序技術,將目標群體基因組序列與已發表的參考基因組進行比對,可以了解目標群體的基因組變異特征,并對群體結構和遺傳多樣性進行分析[15]。為了更好地了解大尾寒羊基因組變異特征和群體結構,本研究對170只大尾寒羊進行全基因組重測序,獲得大尾寒羊基因組SNP、Indel、SV等變異特征,并利用SNP數據對群體遺傳結構和受選擇基因進行了分析,研究結果可為大尾寒羊的保護、開發和利用提供參考,對羊遺傳資源與生物多樣性保護具有重要意義。
1 材料與方法
1.1 試驗動物和體尺測定
選擇位于山東省聊城市國家大尾寒羊保種場的大尾寒羊為研究對象,共采集170只6月齡以上大尾寒羊的血液樣品,其中公羊66只,母羊104只。頸靜脈采血5 mL,EDTA抗凝,帶回實驗室,-20 ℃保存。
依據NY/T 1236—2006《綿山羊生產性能測定技術規范》標準中的方法用羊用測杖和軟尺測量大尾寒羊的體高、體長、胸圍、管圍、臀高、腰高、尾長、尾寬等體尺數據。
1.2 大尾寒羊全血液基因組提取
利用血液基因組DNA提取試劑盒(天根生物,DP348)對每只大尾寒羊血液進行基因組DNA提取。利用10 g·L-1瓊脂糖凝膠電泳檢測提取的基因組DNA完整性,利用微量紫外分光光度計測定DNA濃度和純度。
1.3 大尾寒羊全基因組測序與數據過濾、比對
將合格的DNA樣品送至華大基因生物科技有限公司進行全基因組重測序。簡要步驟如下:將合格的基因組DNA樣品用Covaris儀超聲波隨機打斷為小片段用來構建文庫。之后用Agencourt AMPure XP-Medium kit進行片段選擇,使樣品條帶集中在200~300 bp,并將樣品純化;進行末端修復,加“A”后與接頭連接。對連接產物PCR擴增后進行片段篩選。將PCR產物變性為單鏈后進行環化,完成文庫制備。通過BGISEQ平臺對構建好的文庫進行測序。測序得到的原始圖像數據經base calling轉化為序列數據,即raw reads。用華大基因SOAPnuke (V1.5.6)軟件將原始序列的接頭序列、含N過多和少量低質量序列過濾,得到clean data。應用短序列比對軟件BWA (V0.7.17-r1188)[16]將clean data 比對到參考基因組(GCF_000298735.2_Oar_v4.0_genomic.fna)上,并選擇mapQ值大于30的reads進行后續的變異檢測分析。
1.4 變異檢測
SNP和InDel檢測與注釋:使用GATK (V4.1.2.0)[17]檢測群體所有樣品的SNP和InDel,并對每個變異位點進行質控標記,對于SNP質控條件為“QDlt;2.0FSgt;60.0MQlt;40.0MQRankSumlt;-12.5ReadPosRankSumlt;-8.0”;對于InDel質控條件為“QDlt;2.0 FSgt;200.0 SORgt;10.0MQRankSumlt;-12.5 ReadPosRankSumlt;-8.0”。使用ANNOVAR軟件[18]對每個變異位點進行注釋。結構性變異(structural variation, SV)檢測和注釋:使用Manta (V1.6.0)軟件[19]檢測群體樣品的SV信息,包括插入(insertion, INS)、缺失(deletion, DEL)、染色體易位(translocation)和染色體倒位(inversion)等,使用 ANNOVAR軟件對SV位點進行注釋。拷貝數變異(copy number variation, CNV)檢測與注釋:使用Control-FREEC (V11.6)軟件[20]檢測每個樣品的CNV。根據設定的物種染色體倍性ploidy=2,用Control-FREEC軟件以滑窗的形式自動計算固定長度區域的測序深度,并對其進行標準化,從而得到CNV區域的變異信息。如果連續窗口內的測序深度比該物種染色體倍數大,則表示該區域CNV類型為“gain”,如果連續窗口內的測序深度比該物種染色體倍數小,則該區域CNV類型為“loss”。使用 ANNOVAR軟件對CNV位點進行注釋。
1.5 主成分分析(principal component analysis, PCA)
使用Plink (V1.90)[21]軟件利用SNP標記構建親緣關系G矩陣計算個體間的親緣系數,并計算前3個主成分(PC1、PC2、PC3),結果通過R studio進行可視化。對于親緣關系分析,使用VanRaden算法進行所有樣品之間的親緣關系矩陣計算。
1.6 系統進化樹構建與群體遺傳結構分析
使用FastTree (V2.1.11)軟件[22]構建所有樣品的系統進化樹,使用R包ggtreee對結果進行畫圖。使用Admixture (v1.3)軟件[23]分別將2~8賦值于祖先數目(K值)進行運算,K值的迭代次數依次為10 000,使用R軟件pophelper程序計算K值并對多次重復的結果進行合并,確定最優K值,使用pophelper程序展示其群體結構組成。
1.7 連鎖不平衡分析
使用PopLDdecay (v3.41)[24]軟件計算群體的連鎖不平衡(linkage disequilibrium, LD)衰減率,使用R語言腳本以及Perl語言腳本處理結果繪制LD衰減曲線圖。
1.8 選擇清除分析
使用vcftools (V0.1.16)[25] 軟件,設定窗口大小為100 kb,劃窗距離為50 kb,計算群體內部不同區域的堿基多樣性系數(pi)和偏離中性假說的D系數(Tajima’s D)來篩選受選擇的區域及相關基因。對于受選擇的基因進行GO和KEGG通路富集分析。
2 結 果
2.1 大尾寒羊基因組DNA提取質量檢測
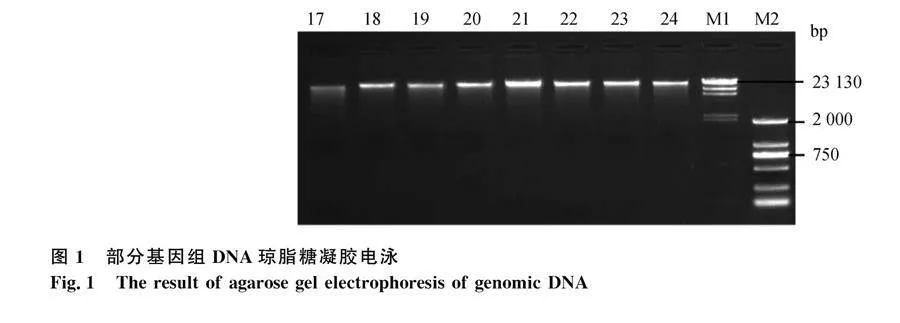
利用瓊脂糖凝膠電泳檢測DNA完整性(圖1),由圖可見DNA條帶單一,表明提取的DNA完整性好,測定濃度和純度后質量合格,可以用于后續分析。
2.2 大尾寒羊全基因組重測序數據質量分析
對170個DNA樣品進行全基因組重測序,共產生1 615.53 G原始測序數據,每只大尾寒羊Raw data平均為9.503 G,Raw reads數量平均值為6.335×107。經過SOAPnuke軟件對數據進行接頭過濾、低質量過濾和去N過濾后,最終得到1 599.56 G高質量的可用數據,平均每只羊獲得的clead data平均值為9.409 G,clean reads平均為6.272×107條。過濾率平均值為99.01%,測序質量較高,Q20%平均為96.35%,Q30%平均為92.23%。用BWA軟件將clean reads比對到參考基因組上,然后使用Qualimap軟件對比對結果進"" 行統計。樣本比對率為98.61%~99.82%,平均測序深度為3.56×(3.01×~3.66×),平均覆蓋度為89.27%~92.14%。
2.3 大尾寒羊變異檢測與注釋
2.3.1 大尾寒羊SNP檢測與注釋
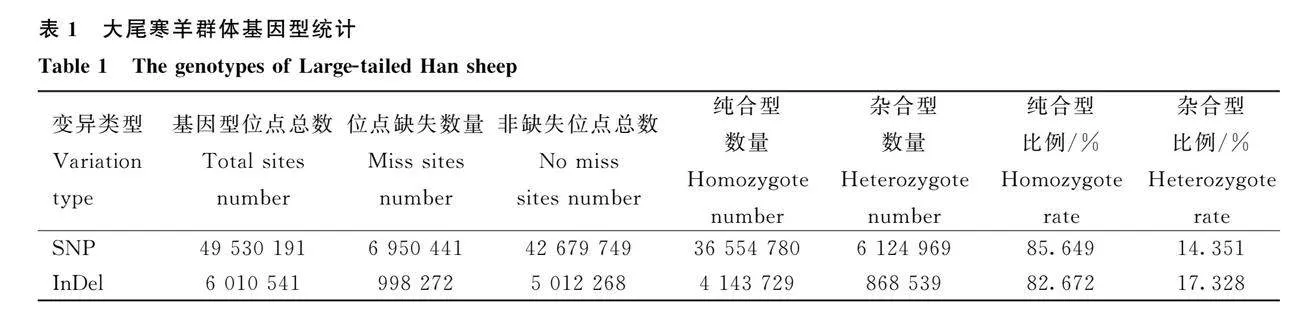
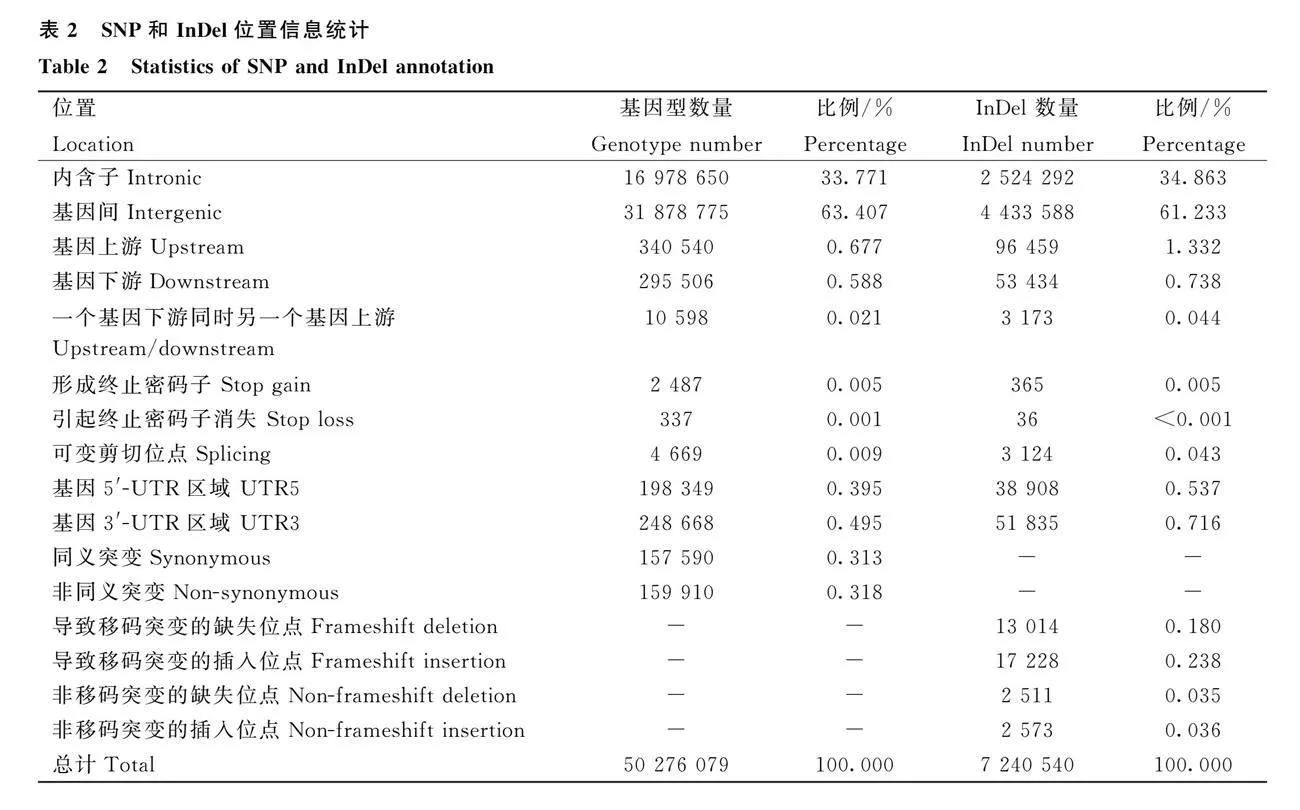
采用GATK3軟件群體call變異的方法,直接基于所有樣品的比對結果進行變異檢測,得到所有樣品的基因型信息。對群體樣品的基因型信息進行統計,結果見表1。
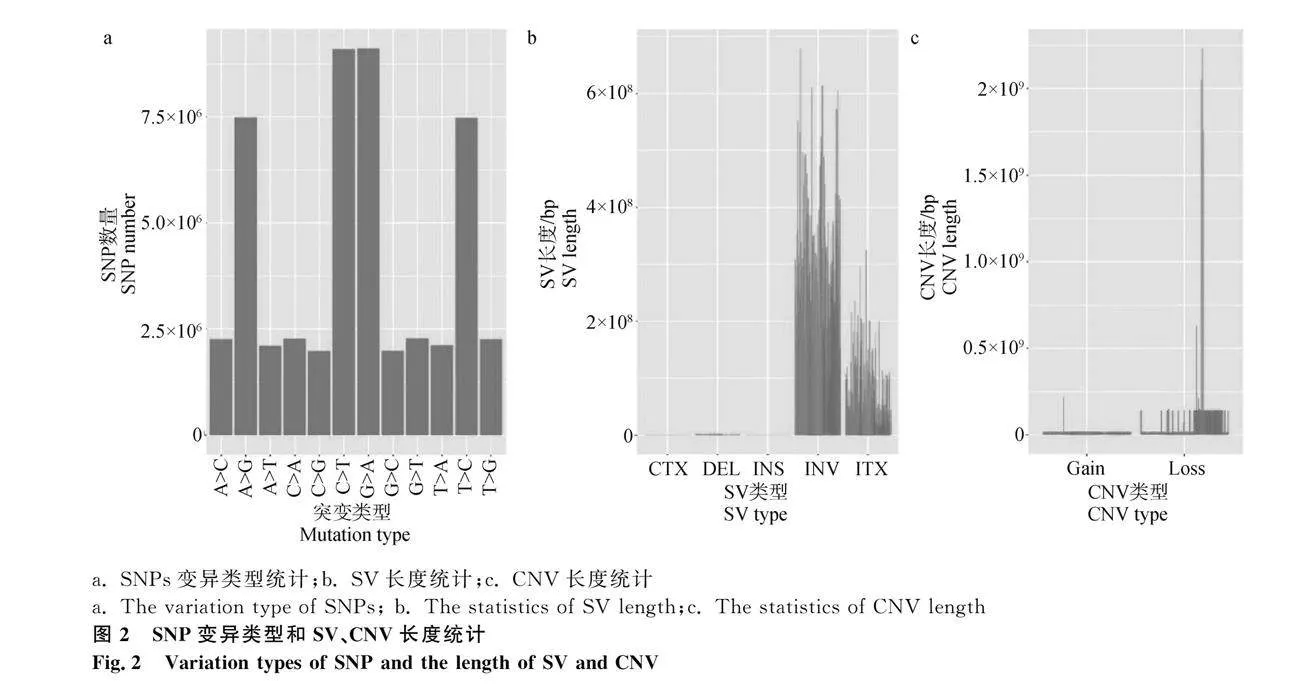
去除缺失位點,共發現42 679 749種基因型,其中純合基因型占85.649%,雜合基因型占14.351%。對每個SNP位點的變異類型進行匯總,如圖2a。在12種SNP變異類型中,Ggt;A、Cgt;T、Agt;G和Tgt;C突變為主要突變類型,Ts/Tv值平均為2.4。同時統計了基因上下游區域、外顯子、內含子和基因間區域的SNPs數量,對位于外顯子區域的突變判斷是否會導致蛋白質序列改變,注釋統計結果見表2。大尾寒羊群體中共發現50 276 079個突變位點,基因間區域和內含子區域的SNP位點數量最多,分別占63.407%和33.771%,其他區域的SNPs不足3%。位于基因內的SNPs中,有2 487個SNPs使終止密碼子提前,編碼蛋白質被截短,而有337個SNP使終止密碼子突變為其他氨基酸密碼子,使編碼的蛋白質長度增加;同時有159 910個SNPs改變了氨基酸編碼;這些使編碼蛋白質發生改變的SNPs是后續重點關注的位點。有447 017個SNPs位于UTR區域,這些SNPs可能對基因表達具有重要的調控作用。
2.3.2 大尾寒羊InDel的檢測與注釋
采用GATK3軟件篩選大尾寒羊基因組中的InDel,基于過濾后的InDel信息,統計樣品的InDel總數、雜合InDel、純合InDel的數量。去除缺失位點后,平均有5 012 268個InDel基因型,其中純合型占82.672%,雜合型占17.328%。對InDel的位置信息進行統計,見表2。同SNP類似,分布在基因間和內含子中的InDel數量最多,占96.096%,其他區域的InDel接近4%。分別有365個和36個InDel使編碼的蛋白質氨基酸數目縮短和增加。有13 014個缺失位點導致蛋白質移碼突變,有17 228個插入位點引起移碼突變。有90 743個InDel位于UTR區域,可能對轉錄后調控有影響。
2.3.3 大尾寒羊SV的檢測與注釋
應用BreakDancer檢測結構性變異,能夠檢測到的結構變異類型主要有插入(INS)、缺失(DEL)、顛換(INV)、染色體內的易位(ITX)、染色體間的易位(CTX)等。對各類型SV數量進行統計,大尾寒羊中CTX數量最多,平均每只羊基因組內有415.82個,其次是DEL和ITX,平均70個。而INS的數量最少,最多的個體有5個INS,平均只有0.25個。與SNP和InDel類似,基因間和基因內SV數量最多,平均有791和181個,外顯子區域平均有44個,而引起剪切位點發生變化的數量較少。分析上述SV長度,INV和ITX長度最大(圖2b)。
2.3.4 大尾寒羊CNV的檢測與注釋
使用control-FREEC軟件檢測每個樣品的CNV,劃窗長度為50 kb,分別對CNV偏高(gain)和偏低(loss)的區域數量進行統計,與其他類型變異不同,CNV數量最多的區域是外顯子(平均175個)和基因間(平均83個),而其他區域CNV數量都較少。統計發生拷貝數變異片段的長度,拷貝數異常減少的片段長度要明顯比異常增多的片段長度大,有些個體發生了大片段拷貝數減少(圖2c)。
2.4 大尾寒羊群體結構分析
2.4.1 群體主成分分析
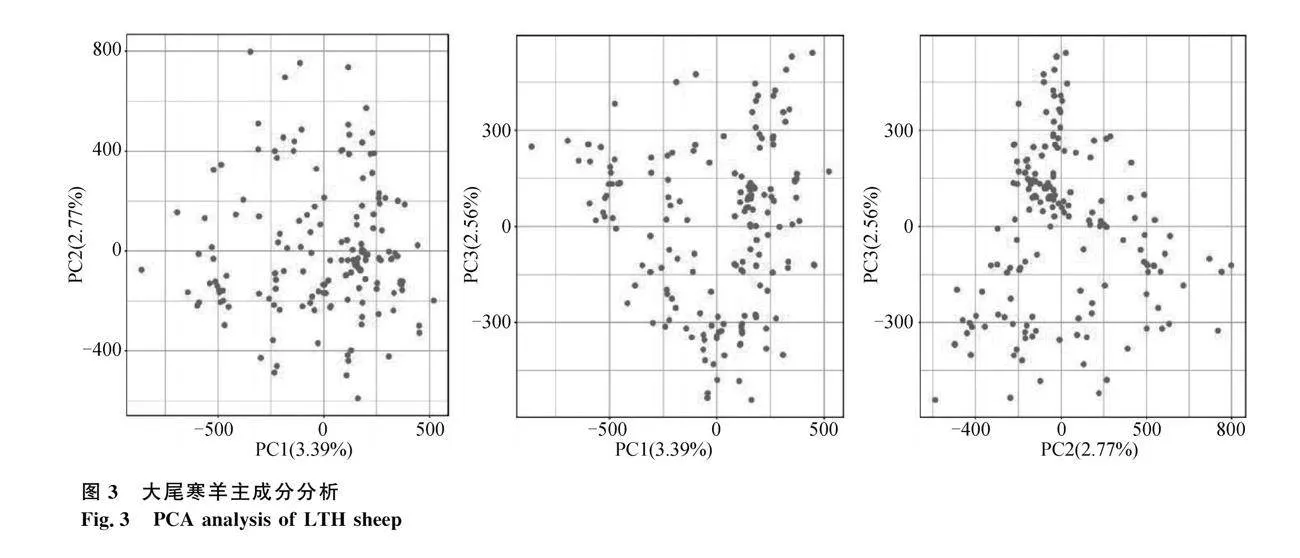
對大尾寒羊170個個體進行主成分分析,結果(圖3)表明,主成分1能解釋3.39%的變異,主成分2能解釋2.77%的變異,主成分3能解釋2.56%的變異。大尾寒羊群內個體較為分散,有一些個體聚集,但聚集不集中且不明顯。
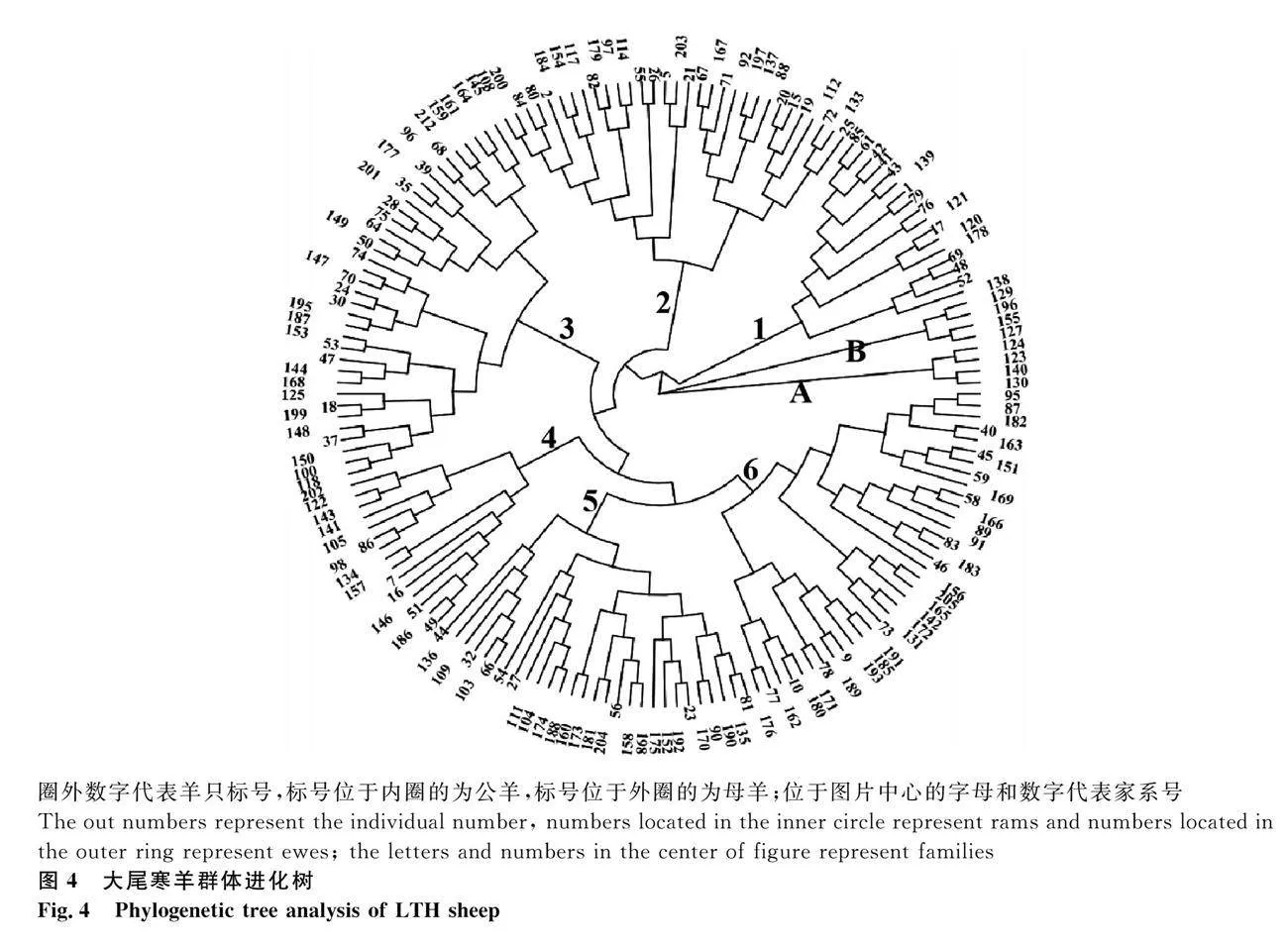
2.4.2 群體進化樹與群體遺傳結構分析
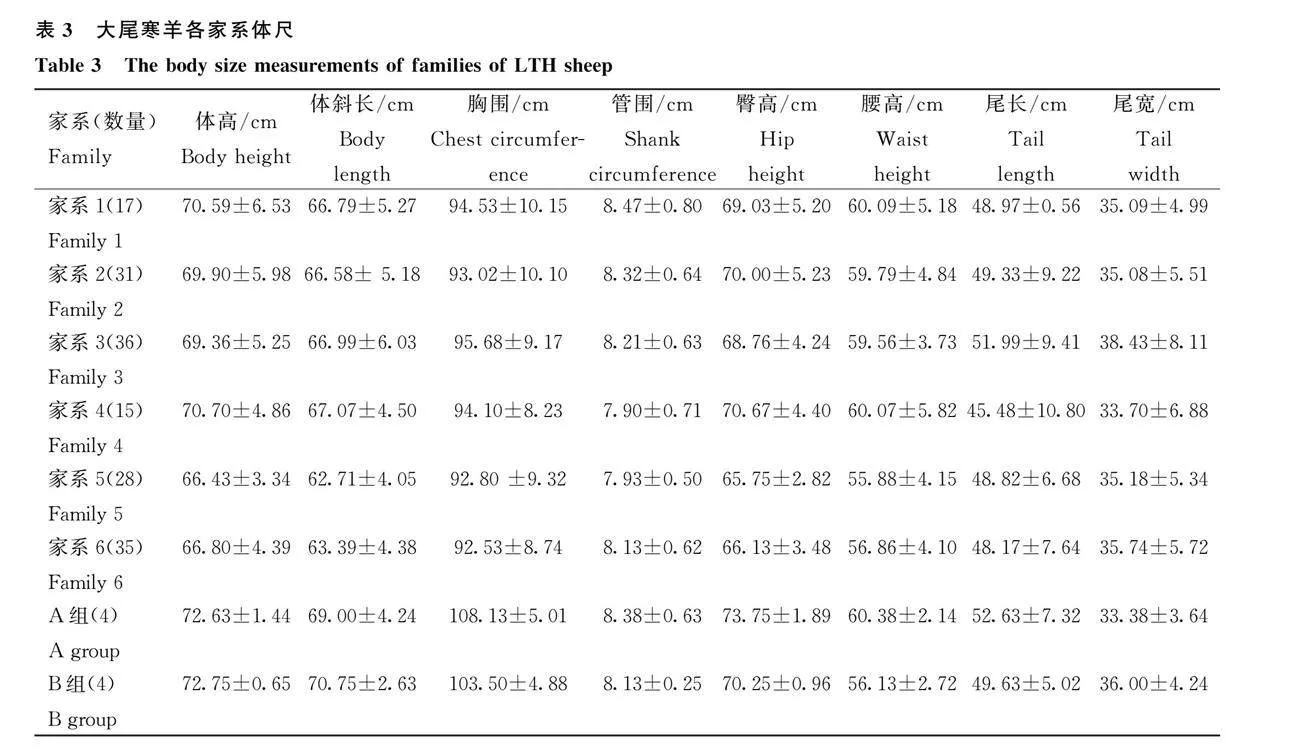
用FastTree軟件構建所有大尾寒羊樣品的最大似然法系統發育樹,見圖4。根據系統發育樹結果,可以將大尾寒羊分為6個家系,不同家系個體數量差別較大,家系3含36個個體,而家系4只含15個個體。另外有8只母羊(圖4中A組和B組)與公羊親緣關系較遠。根據家系將大尾寒羊6個家系和8只親緣關系較遠母羊的體尺數據進行整理,見表3。不同家系間體尺數據比較發現家系5和家系6的體高、體長、臀高和腰高小于其他4個家系,體型偏小;而家系4的尾長和尾寬小于其他5個家系。A組和B組各有4只母羊,雖然是母羊但它們的體高、體長和胸圍均較大。
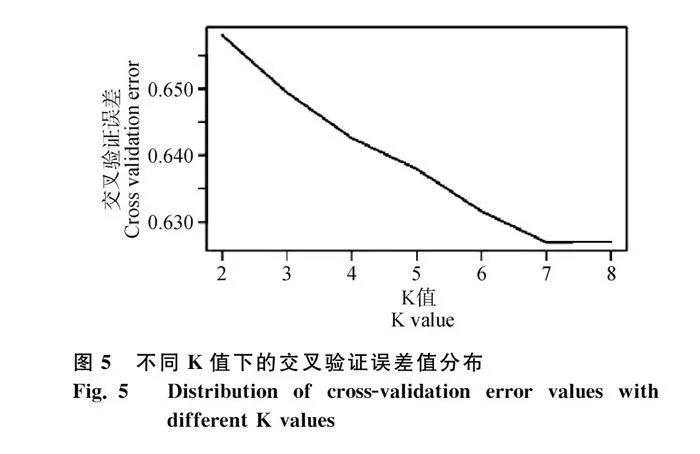
應用Admixture軟件對所有樣品進行聚類,計算相應的Q值(第i個樣品其基因組變異源于第k群體的概率),并繪制群體結構圖,以確定每個樣品的遺傳組成。利用交叉驗證誤差圖方法對K值分析,當K=7時為假定的最佳分群數目(圖5)。
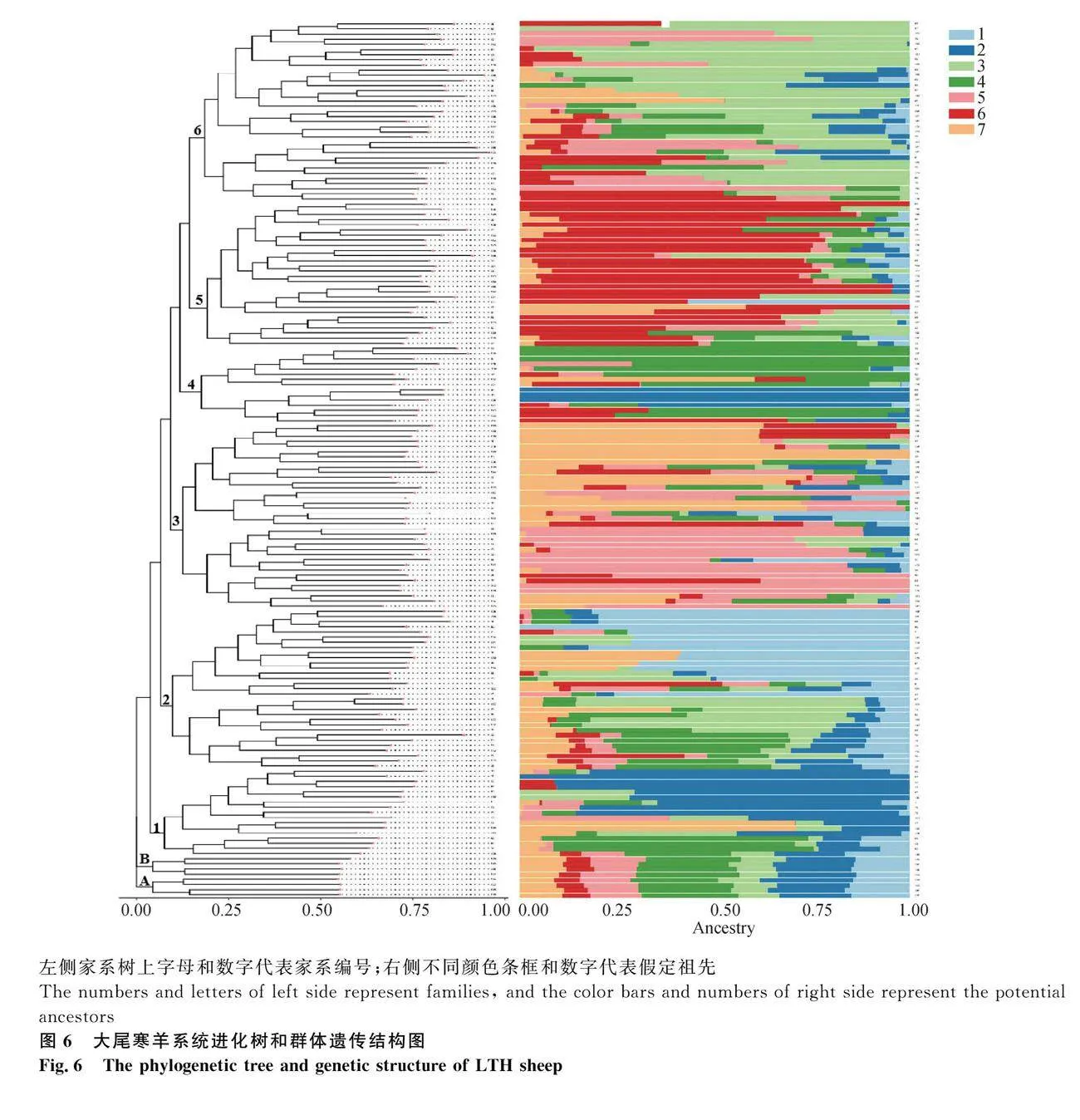
將進化樹和群體結構圖(K=7時)相結合,繪制系統進化樹和群體遺傳結構圖(圖6)。從圖中可以看出,有些個體具有較為單一的祖先成分,如母羊87幾乎只有祖先1成分,公羊61、母羊86、98和105幾乎只有祖先2成分,公羊80幾乎只有祖先3成分,母羊186、公羊49和51幾乎只有祖先4 成分,母羊159、212和187的祖先5成分占99.99%,公羊81祖先成分的99.99%是祖先6,公羊18和母羊199幾乎只有祖先7成分。
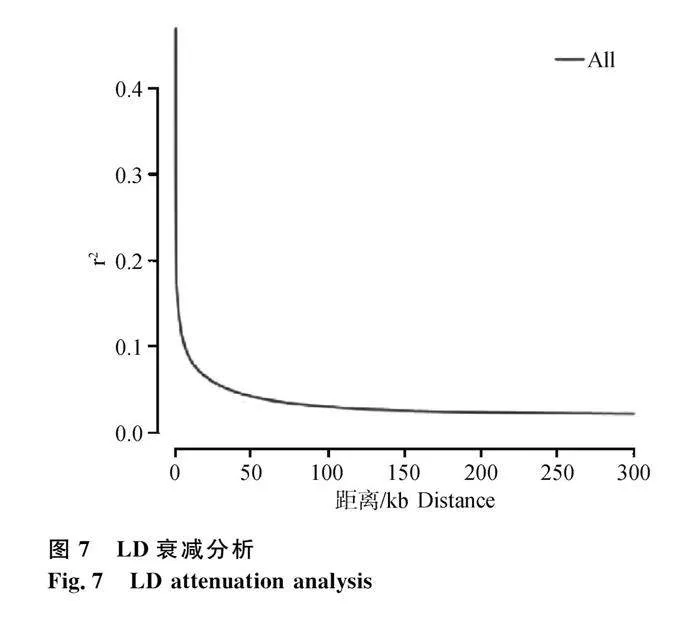
2.4.3 群體連鎖不平衡分析" 使用PopLDDecay軟件對群體進行LD分析,利用LD衰減分析群體的連鎖不平衡狀態(圖7),大尾寒羊群體的SNP距離在0~50 kb范圍內衰減速度從0.51下降至到0.04,LD衰減系數從0.51衰減到0.1的距離只有5.2 kb,衰減速度快,表明該大尾寒羊群體遺傳多樣性相對較高。
2.5 大尾寒羊基因組選擇清除分析
利用vcftools軟件分析確定了72個受選擇的基因,如ALOX12B、ALOX15B、AWAT1、SREBF1、GBA2、CERS5和CREB3等,對這些基因進行GO和KEGG分析。其中顯著富集的GO條目主要有亞油酸代謝過程(linoleic acid metabolic process)、花生四烯酸代謝過程(arachidonic acid metabolic process )、長鏈脂肪酸合成和代謝過程(long-chain fatty acid biosynthetic and metabolic process)、肝氧蛋白生物合成和代謝過程(hepoxilin "biosynthetic and metabolic process)和黏著斑蛋白結合(vinculin binding)等。顯著富集的KEGG信號通路有胰島素分泌(insulin secretion)、花生四烯酸代謝(arachidonic acid metabolism)、鞘脂代謝(sphingolipid metabolism)、胰高血糖素信號通路(glucagon signaling pathway)、產熱(thermogenesis)等信號通路。
3 討 論
大尾寒羊是長脂尾綿羊品種,是我國非常寶貴的羊遺傳資源。近幾十年來,與蘭州大尾羊[26]、廣靈大尾羊[27]等長脂尾羊一樣,由于雜交等多種原因大尾寒羊數量銳減。為了防止這一優良品種和基因流失,國家和山東省均已建立了保種場,并進行了多種形式的保種,取得了一定的成果。但與其他肉用綿羊品種相比,大尾寒羊產肉性能競爭力較弱,其重要產品尾脂目前利用價值較低,加上對大尾寒羊的開發和利用進展較為緩慢,使大尾寒羊保種仍面臨諸多困難和挑戰。為了全面了解大尾寒羊現存群體結構特征,本研究利用全基因組重測序對大尾寒羊基因組變異包括SNP、InDel、SV和CNV進行了分析,并依據SNP信息對大尾寒羊親緣關系、群體結構、連鎖不平衡衰減特征進行了分析,為大尾寒羊保種工作提供依據。
基于全基因組重測序數據分析群體遺傳結構是評估地方品種群體情況的有效手段[28]。目前已有不少研究對不同綿羊品種如皮山紅羊[29]、永登七山羊[30]、新疆細毛羊[31]、祁連白藏羊[32]、蒙古羊和巴盟肉羊[33]等進行了全基因組重測序和群體遺傳結構分析,為這些品種的開發和利用提供了有效信息。本研究采集了保種場內6月齡及以上全部大尾寒羊個體樣品,去除有明確系譜記錄的羊,剩余170只進行了全基因組重測序。對測序數據質控后,進行了遺傳變異分析。在12種SNPs變異類型中,Ggt;A和Cgt;T突變數量最多,且遠多于其他突變類型,Ts與Tv比例平均為2.4,與其他綿羊群體接近[34-35]。大尾寒羊中發現了50 276 079個SNPs,比其他綿羊品種如涼山半細毛羊[36]、東北細毛羊[37]和一些國外品種[38]數量多。大尾寒羊中發現了7 240 540個InDel,也比涼山半細毛羊[36]多。大尾寒羊群體內遺傳變異較豐富,是重要的遺傳資源,建議加大對大尾寒羊的保護力度。在大尾寒羊群體中SNPs突變主要位于基因間區域(63.41%),其次是內含子區域(33.77%),與東北細毛羊類似[28],與涼山半細毛羊[36]差別較大,而Sun等[36]在涼山半細毛羊中發現的SNPs主要位于內含子區域(62.94%),其次是基因間區域(25.12%)。大尾寒羊基因組結構性變異中染色體間的易位(CTX)數量最多,INS數量最少,而其他4個綿羊品種(南丘綿羊、高山美利奴綿羊、祁連白藏羊和歐拉羊)中DEL和INS最多,但該研究中未分析CTX變異[37]。這些結果表明,大尾寒羊基因組變異較為豐富,另外群體連鎖不平衡分析也表明大尾寒羊群體遺傳多樣性較高,與劉澤民等[39]用微衛星和線粒體D-loop區聯合分析得出的結論一致。近十幾年來,隨著人們消費習慣和羊飼養模式的改變,大尾寒羊的長脂尾不再受人們喜愛,數量銳減,處于保種狀態,目前大尾寒羊的育種工作開展較少,也可能是其群體遺傳多樣性相對豐富的原因之一。但目前大尾寒羊群體數量少,盡管含有較高的遺傳多樣性,保種過程中應盡可能避免遺傳距離較近的個體間的交配。
利用PCA分析可以了解群體遺傳結構,個體遺傳關系越近,PCA圖上顯示的直線距離就越近,反之,直線距離就越遠。對大尾寒羊群體進行PCA分析,發現群體內整體較為分散,有一些個體聚集,表明大尾寒羊群體可能并未受到某種強烈選擇,也可能因為是同一個品種樣本較多。對大尾寒羊群體構建系統發育樹,可以將大尾寒羊分為6個家系,但每個群體數量差異較大,在后續保護過程中,要注意對小家系進行擴繁。根據各家系體尺數據分析結果(表3),家系5和家系6的體型比其他4個家系偏小,家系4的尾比其他家系小。根據這些特點,可以按照大體型、小體型、小型尾等方向進行定向選育。另外,可以從河南省引進大尾寒羊進行雜交,但要制定科學合理的雜交計劃,在保證山東大尾寒羊純度的基礎上,豐富大尾寒羊的遺傳多樣性。利用新技術建立大尾寒羊胚胎和體細胞保存技術流程,對各個家系進行胚胎保存,對已經保存的精液定期檢測,及時更新和補充。另外有8只母羊與這6個家系親緣關系較遠,可以引入外源公羊,充分利用這8只母羊增加家系數量。對群體進行祖先成分分析,K=7時為最佳分群數目。對個體的祖先成分分析,有不少個體幾乎只含有一個祖先成分,對于這些個體尤其是公羊,在后期保護和利用過程中要重點關注。
對大尾寒羊基因組進行選擇清除分析,受選擇基因富集的GO條目和KEGG信號通路主要是一些與脂質代謝相關的信號通路。一些對脂尾羊和瘦尾羊的尾脂轉錄組、脂質組學等研究中都有富集到與脂質代謝相關的信號通路[4-5,7,40-41]。本研究中也富集到了與胰高血糖素、胰島素相關的信號通路。胰高血糖素和胰島素是調控能量代謝的重要激素,不同尾型的綿羊脂肪組織的轉錄組分析中也富集到相關信號通路[5,42]。大尾寒羊屬長脂尾羊,其尾部脂肪在冬季寒冷時產熱以維持體溫,在秋季又開始沉積脂肪,在脂肪沉積與分解的動態過程中這些與脂肪代謝相關的基因起著重要作用。在長期循環過程中,這些與脂肪代謝、產熱等相關基因受到強烈選擇,它們特異的時空表達賦予長脂尾綿羊獨特的脂肪代謝特征,利于它們能夠在惡劣的環境中生存。
4 結 論
本研究基于全基因組重測序分析了170只大尾寒羊的基因組變異特征和群體結構,結果顯示大尾寒羊群體內具有較為豐富的遺傳變異,群體遺傳多樣性較為豐富。根據親緣關系將群體劃分為6個家系,群體遺傳多樣性較豐富,但家系數量較少,需要制定合理的配種計劃,同時應注重大尾寒羊的開發和利用。
參考文獻(References):
[1] 國家畜禽遺傳資源委員會.中國畜禽遺傳資源志(羊志)[M].北京:中國農業出版社,2011.
China National Commission of Animal Genetic Resources.Animal genetic resources in China: sheep and goats[M].Beijing: China Agriculture Press,2011.(in Chinese)
[2] KALDS P,LUO Q,SUN K,et al.Trends towards revealing the genetic architecture of sheep tail patterning: promising genes and investigatory pathways[J].Anim Genet,2021,52(6):799-812.
[3] FARHADI S,HASANPUR K,GHIAS J S,et al.Comprehensive gene expression profiling analysis of adipose tissue in male individuals from fat-and thin-tailed sheep breeds[J].Animals (Basel),2023,13(22):3475.
[4] BAKHTIARIZADEH M R.Deciphering the role of alternative splicing as a potential regulator in fat-tail development of sheep:a comprehensive RNA-seq based study[J].Sci Rep,2024,14(1):2361.
[5] XU Y X,WANG B,JING J N,et al.Whole-body adipose tissue multi-omic analyses in sheep reveal molecular mechanisms underlying local adaptation to extreme environments[J].Commun Biol,2023,6(1):159.
[6] HOU M,YE M J,MA X L,et al.Colon microbiota and metabolite potential impact on tail fat deposition of Altay sheep[J]. Microbiol Spectr,2024,12(6):e0310323.
[7] CAIYE Z,SONG S Z,LI M N,et al.Genome-wide DNA methylation analysis reveals different methylation patterns in Chinese indigenous sheep with different type of tail[J].Front Vet Sci,2023,10:1125262.
[8] LI T T,JIN M L,WANG H H,et al.Whole-genome scanning for selection signatures reveals candidate genes associated with growth and tail length in sheep[J].Animals (Basel),2024,14(5):687.
[9] DENG J,XIE X L,WANG D F,et al.Paternal origins and migratory episodes of domestic sheep[J].Curr Biol,2020,30(20): 4085-4095.e6.
[10] ZHU C Y,LI N,CHENG H P,et al.Genome wide association study for the identification of genes associated with tail fat deposition in Chinese sheep breeds[J].Biol Open,2021,10(5):bio054932.
[11] YUAN Z,LIU E,LIU Z,et al.Selection signature analysis reveals genes associated with tail type in Chinese indigenous sheep[J].Anim Genet,2017,48(1):55-66.
[12] 章 煥,張 成,邸騰剛,等.大尾寒羊和小尾寒羊尾部脂肪lncRNA差異表達分析[J].河南農業大學學報,2023, 57(2): 298-306.
ZHANG H,ZHANG C,DI T G,et al.Differential expression analysis of tail fat LncRNA in Large-tailed Han sheep and Small-tailed Han sheep breeds[J].Journal of Henan Agricultural University,2023,57(2):298-306.(in Chinese)
[13] 楊廣禮,劉凱迪,付歡歡,等.大尾寒羊和小尾寒羊尾脂、心脂和腎脂脂肪細胞比較研究[J].黑龍江畜牧獸醫, 2019(19): 60-64,179.
YANG G L,LIU K D,FU H H,et al.Comparative study on adipocytes of tail fat, pericardiac fat and perirenal adipose tissue of Large-tailed Han sheep and Small-tailed Han sheep breeds[J].Heilongjiang Animal Science and Veterinary Medicine, 2019(19):60-64,179.(in Chinese)
[14] 張果平,蔡中峰.山東省綿羊、山羊種業發展現狀、問題與建議[J].山東畜牧獸醫,2022,43(4):72-76.
ZHANG G P,CAI Z F.The development status,problem and suggestion on sheep and goat breeding industry in Shandong Province[J].Shandong Journal of Animal Science and Veterinary Medicine,2022,43(4):72-76.(in Chinese)
[15] IANNUCCI A,BENAZZO A,NATALI C,et al.Population structure, genomic diversity and demographic history of Komodo dragons inferred from whole-genome sequencing[J].Mol Ecol,2021,30(23):6309-6324.
[16] LI H,DURBIN R.Fast and accurate short read alignment with Burrows-Wheeler transform[J].Bioinformatics,2009,25(14): 1754-1760.
[17] MCKENNA A,HANNA M,BANKS E,et al.The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data[J].Genome Res,2010,20(9):1297-1303.
[18] WANG K,LI M Y,HAKONARSON H.ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data[J].Nucleic Acids Res,2010,38(16):e164.
[19] CHEN X Y,SCHULZ-TRIEGLAFF O,SHAW R,et al.Manta:rapid detection of structural variants and indels for germline and cancer sequencing applications[J].Bioinformatics,2016,32(8):1220-1222.
[20] BOEVA V,POPOVA T,BLEAKLEY K,et al.Control-FREEC: a tool for assessing copy number and allelic content using next-generation sequencing data[J].Bioinformatics,2012,28(3):423-425.
[21] PURCELL S,NEALE B,TODD-BROWN K,et al.PLINK: a tool set for whole-genome association and population-based linkage analyses[J].Am J Hum Genet,2007,81(3):559-575.
[22] PRICE M N,DEHAL P S,ARKIN A P.FastTree 2-approximately maximum-likelihood trees for large alignments[J].PLoS One,2010,5(3):e9490.
[23] ALEXANDER D H,NOVEMBRE J,LANGE K.Fast model-based estimation of ancestry in unrelated individuals[J].Genome Res,2009,19(9):1655-1664.
[24] ZHANG C,DONG S S,XU J Y,et al.PopLDdecay: a fast and effective tool for linkage disequilibrium decay analysis based on variant call format files[J].Bioinformatics,2019,35(10):1786-1788.
[25] DANECEK P,AUTON A,ABECASIS G,et al.The variant call format and VCFtools[J].Bioinformatics,2011,27(15): 2156-2158.
[26] 陳宏福,韋 體,張俊松,等.國家級畜禽遺傳資源蘭州大尾羊保護現狀與對策[J].甘肅畜牧獸醫,2023,53(1):15-18.
CHEN H F,WEI T,ZHANG J S,et al.The conservational status and recommendation of the national livesrock and poultry genetic resource-Lanzhou Large-tailed sheep[J].Gansu Animal Husbandry and Veterinary Medicine,2023,53(1):15-18.(in Chinese)
[27] 高 毅,鞏薇娜,張明娟,等.淺析廣靈大尾羊資源保護[J].中國畜牧業,2023(10):53-54.
GAO Y,GONG W N,ZHANG M J,et al.General views on the conservation of Guangling Large-tailed sheep[J].China Animal Industry,2023(10):53-54.(in Chinese)
[28] 趙真堅,王書杰,陳 棟,等.基于低深度全基因組測序分析內江豬群體結構和遺傳多樣性[J].畜牧獸醫學報,2023, 54(6): 2297-2307.
ZHAO Z J,WANG S J,CHEN D,et al.Population structure and genetic diversity analysis of Neijiang pigs based on low-coverage whole genome sequencing[J].Acta Veterinaria et Zootechnica Sinaica,2023,54(6):2297-2307.(in Chinese)
[29] 石 蘭,馬梅蘭,木合塔帕·買買提江,等.基于全基因組重測序解析皮山紅羊群體遺傳結構及產羔數候選基因研究[J].中國畜牧獸醫,2024,51(2):624-638.
SHI L,MA M L,MUHETAPA M M T J,et al.Study on the genetic structure and litter size candidate genes of Pishan Red sheep population based on whole genome resequencing[J].China Animal Husbandry amp; Veterinary Medicine,2024,51(2):624-638.(in Chinese)
[30] 馬克巖,韓金濤,白雅琴,等.基于簡化基因組測序的永登七山羊遺傳多樣性分析[J].畜牧獸醫學報,2023,54(5):1939-1950.
MA K Y,HAN J T,BAI Y Q,et al.Genetic diversity analysis of Yongdeng Qishan sheep based on specific-locus amplified fragment sequencing[J].Acta Veterinaria et Zootechnica Sinica,2023,54(5):1939-1950.(in Chinese)
[31] 陳開旭,郭翠潔,楊 帆,等.基于全基因組重測序分析新疆細毛羊遺傳多樣性[J].新疆農業科學,2023, 60(5):1292-1300.
CHEN K X,GUO C J,YANG F,et al.Genetic diversity analysis of Xinjiang sheep with fine wool based on whole-genome re-sequencing[J].Xinjiang Agricultural Sciences,2023,60(5):1292-1300.(in Chinese)
[32] 余道寧,王 佟,鐵中華,等.基于全基因組重測序對祁連白藏羊類群的群體結構分析[J].中國草食動物科學,2023, 43(6): 12-16.
YU D N,WANG T,TIE Z H,et al.Population structure analysis of Qilian White Tibetan sheep group based on whole genome resequencing[J].China Herbivore Science,2023,43(6):12-16.(in Chinese)
[33] 常晨城,白音巴圖,周 樂,等.內蒙古地方綿羊遺傳多樣性及尾椎數性狀相關選擇信號研究[J].中國畜牧雜志,2023, 59(12): 109-115.
CHANG C C,BAIYINBATU,ZHOU L,et al.Analysis on the genetic diversity and selection signatures for the caudal vertebrae number of local sheep species of Inner Mongolia,China[J].Chinese Journal of Animal Science,2023,59(12):109-115.(in Chinese)
[34] LI J,XU H,LIU X F,et al.Insight into the possible formation mechanism of the intersex phenotype of Lanzhou Fat-tailed Sheep using whole-genome resequencing[J].Animals (Basel),2020,10(6):944.
[35] YI W F,HU M Y,SHI L L,et al.Whole genome sequencing identified genomic diversity and candidated genes associated with economic traits in Northeasern Merino in China[J].Front Genet,2024,15:1302222.
[36] SUN X L,GUO J Z,LI R,et al.Whole-genome resequencing reveals genetic diversity and wool trait-related genes in Liangshan semi-fine-wool sheep[J].Animals (Basel),2024,14(3):444.
[37] LI R N,ZHAO Y H T,LIANG B M,et al.Genome-wide signal selection analysis revealing genes potentially related to sheep-milk-production traits[J].Animals (Basel),2023,13(10):1654.
[38] QIAO G Y,XU P,GUO T T,et al.Genome-wide detection of structural variation in some sheep breeds using whole-genome long-read sequencing data[J].J Anim Breed Genet,2024,141(4):403-414.
[39] 劉澤民,王巖超,張淑二,等.利用微衛星和線粒體D-loop區聯合分析大尾寒羊的遺傳多樣性[J].山東畜牧獸醫,2019, 40(7): 1-5.
LIU Z M,WANG Y C,ZHANG S E,et al.Analysis on the genetic diversity of Large-tailed Han sheep using mtDNA D-loop and microsatellite loci[J].Shandong Journal of Animal Science and Veterinary Medicine,2019,40(7):1-5.(in Chinese)
[40] BAKHTIARIZADEH M R,SALEHI A,ALAMOUTI A A,et al.Deep transcriptome analysis using RNA-Seq suggests novel insights into molecular aspects of fat-tail metabolism in sheep[J].Sci Rep,2019,9(1):9203.
[41] HE X G,WU R H,YUN Y Y,et al.MicroRNA and circular RNA profiling in the deposited fat tissue of Sunite sheep[J].Front Vet Sci,2022,9:954882.
[42] LIU T Y,FENG H,YOUSUF S,et al.Genome-wide analysis of microRNAs identifies the lipid metabolism pathway to be a defining factor in adipose tissue from different sheep[J].Front Vet Sci,2022,9:938311.
(編輯 郭云雁)
