葛根芩連湯數字化色譜指紋譜的建立及其與組方藥味的相關性研究
2010-01-30 06:23:26姚亞敏王新宏
中成藥 2010年10期
安 叡, 姚亞敏, 王新宏
(1.上海中醫藥大學中藥學院,上海201203;2.上海市公共衛生臨床中心復旦大學附屬公共衛生臨床中心,上海201508)
葛根芩連湯為東漢醫圣張仲景名方,出自《傷寒論·太陽篇》,由葛根、黃芩、黃連、炙甘草四味中藥組成,有解表清里,升陽止瀉功效,用于瀉熱下利[1]。指紋圖譜作為控制中藥復方質量的重要而有效的手段,與中藥復方有效成分的復雜性、不確知性,以及發揮作用時眾多成分的協同性、多靶點性特點相吻合[2]。但僅通過色譜方法得到的中藥樣品色譜圖應該僅為樣品的化學輪廓圖,必須通過一定的數據處理和多個相關樣品比較,指紋峰的歸屬鑒別等后才可以賦予色譜圖以特征性。本研究采用梯度洗脫技術進行HPLC分離分析,建立葛根芩連湯的數字化色譜指紋譜,并分別對方中葛根、黃芩、黃連進行色譜指紋譜鑒別,探求各藥材所呈現的特征峰群,建立復方與藥材化學組成之間的聯系,有助于提高中藥復方質量控制水平。希望為闡明葛根芩連湯藥效物質基礎,從化學成分分析其配伍規律奠定基礎。
1 儀器與試藥
Agilent 1100型高效液相色譜儀(G1379A Degasser;G1311A Quantum;G1316A Column;G1314A Vwd);SK3200H超聲儀(上海科導超聲儀器有限公司)。
葛根、黃芩、黃連和炙甘草藥材均購于上海康橋中藥飲片有限公司,經本校趙志禮教授鑒定。
葛根素、大豆苷、大豆苷元、黃芩苷、黃芩素、漢黃芩素、鹽酸小檗堿、鹽酸巴馬汀對照品(批號分別為110752-200511、111738-200501、111502-200101、 110715-200815、111595-200604、 111514-200403、 110713-200609、 110732-200506)均購自中國藥品生物制品檢定所;漢黃芩苷,批號061013;購自上海友思生物技術有限公司。
甲醇(HPLC級購于國藥集團化學試劑有限公司),甲酸、三乙胺等試劑為分析純,水為重蒸水。
2 方法與結果
2.1 色譜條件 色譜柱:Agilent TC-C18(4.6 mm×250 mm,5 μm);流動相:甲醇-含0.5%甲酸和0.5%三乙胺的水溶液;梯度:在0~90 min內甲醇從26%線性升至70%;流速:1 mL/min;檢測波長:270 nm;柱溫:30 ℃;進樣量:5 μL。
2.2 溶液的配制
2.2.1 復方供試品溶液的制備 按處方配比精密取葛根芩連湯各味藥飲片(葛根約15 g,黃芩約9 g,黃連約9 g,甘草約6 g),加10倍量水煎煮45 min,濾過,再加8倍量水煎煮30 min,減壓回收干燥,得提取物。取提取物約0.5 g,精密稱定,加甲醇25 mL,超聲提取后經0.45 μm微孔濾膜濾過,得復方供試品溶液。
2.2.2 藥材供試品溶液的制備 按處方量各取葛根、黃芩、黃連藥材,精密稱定,按復方相同制備方法處理,得各藥材提取物,取葛根藥材提取物約2 mg、黃芩藥材提取物約1 mg、黃連藥材提取物約1 mg,精密稱定,各加甲醇10 mL,經0.45 μm微孔濾膜濾過,得各藥材供試品溶液。
2.2.3 陰性供試品溶液的制備 稱取除某味藥材的陰性處方,按復方相同制備方法處理,得各陰性樣品提取物,各取葛根、黃芩、黃連陰性提取物約3 mg,精密稱定,加甲醇10 mL,經0.45 μm微孔濾膜濾過,得各陰性供試品溶液。
2.2.4 對照品溶液配制 精密稱取經五氧化二磷干燥48 h的各對照品適量,用甲醇溶解并定容,制備各對照品的單標對照品溶液:葛根素濃度為0.35 mg/mL、大豆苷濃度為0.13 mg/mL、大豆苷元濃度為1.02 mg/mL、黃芩苷濃度為0.0712 mg/mL,漢黃芩苷濃度為0.18 mg/mL,黃芩素濃度為0.36 mg/mL,漢黃芩素濃度為0.14 mg/mL,鹽酸小檗堿濃度為0.46 mg/mL,鹽酸巴馬汀濃度為0.52 mg/mL,備用。
2.3 指紋圖譜方法學考察
2.3.1 對照品的檢出 在確定的色譜條件下對葛根芩連湯中的葛根素、大豆苷、大豆苷元、黃芩苷、黃芩素、漢黃芩苷、漢黃芩素、鹽酸小檗堿、鹽酸巴馬汀均能檢出,峰形均較理想(見圖1)。
2.3.2 精密度試驗 取同一供試品溶液,按上述色譜條件連續進樣5次,記錄各組分的保留時間和峰面積,各組分保留時間的RSD均小于0.71%,各組分峰面積的RSD均小于0.58%,結果表明儀器精密度良好。
2.3.3 穩定性試驗 取同一供試品溶液,分別于0、2、4、6、12、24、36 h在上述色譜條件下進樣分析,記錄各組分的保留時間和峰面積,各組分保留時間的RSD均小于1.29%,各色譜峰峰面積的RSD均小于0.98%,結果表明供試品溶液在36h內穩定(見圖2)。
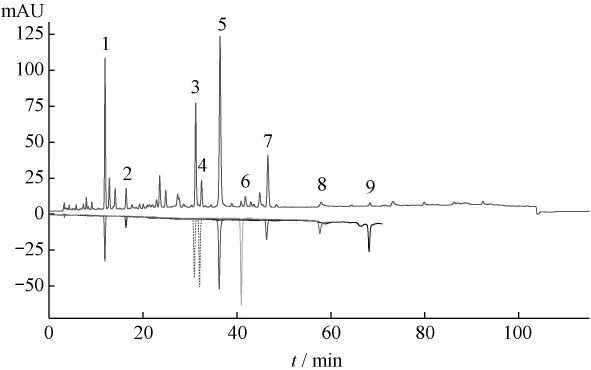
圖1 葛根芩連湯HPLC圖中對照品的檢出
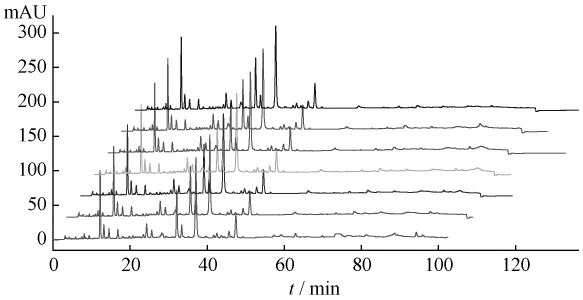
圖2 不同時間間隔測定所得HPLC圖
2.3.4 重復性試驗 按處方配比取葛根芩連湯各味藥飲片,各取5份,按2.2.1項方法制備樣品,在上述色譜條件下進樣分析,記錄各組分的保留時間和峰面積,各組分保留時間的RSD均小于1.41%,各組分峰面積的 RSD均小于1.71%,結果表明本試驗重復性良好。
2.4 HPLC相對保留值指紋譜的建立 本研究采用HPLC技術對葛根芩連湯及葛根、黃芩、黃連藥材一次進樣進行梯度分離分析,得到眾多的色譜峰(見圖3~5),利用相對保留值(α)及樣品峰相對面積值(RA)組成各樣品的HPLC數字化色譜指紋譜(HPLC-FPS)[3-6],通過與陰性樣品比較,可找出葛根芩連湯中用以鑒定上述藥材的較全面的特征峰群,實現對制劑中藥材的鑒定。表1為以葛根素為內參照峰的葛根藥材及樣品的色譜指紋譜的特征峰;表2為以黃芩苷為內參照峰的黃芩藥材及樣品的色譜指紋譜的特征峰;表3為以鹽酸小檗堿為內參照峰的黃連藥材及樣品的色譜指紋譜的特征峰。
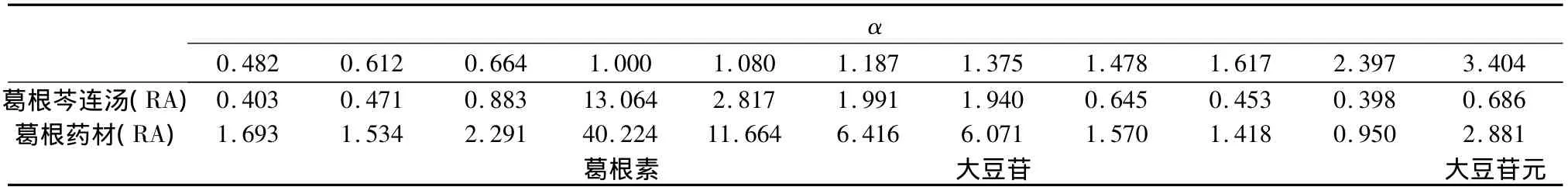
表1 葛根藥材在樣品中的特征峰
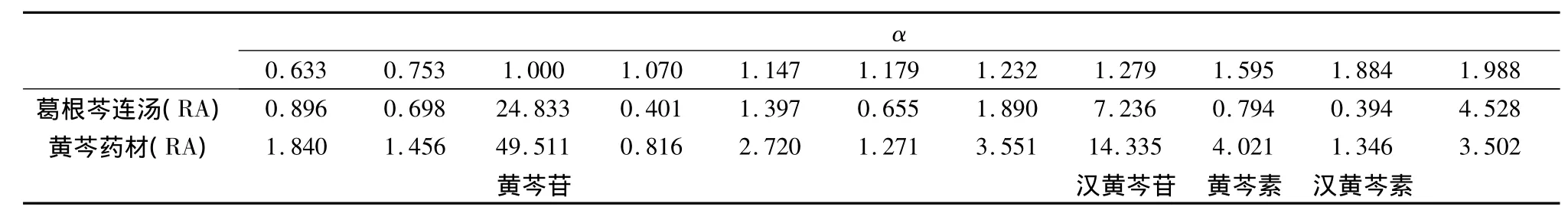
表2 黃芩藥材在樣品中的特征峰
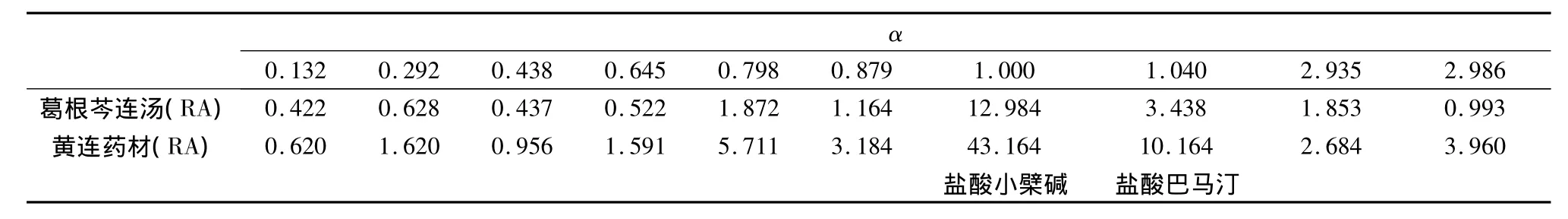
表3 黃連藥材在樣品中的特征峰

圖3 葛根芩連湯、葛根藥材及陰性樣品HPLC圖
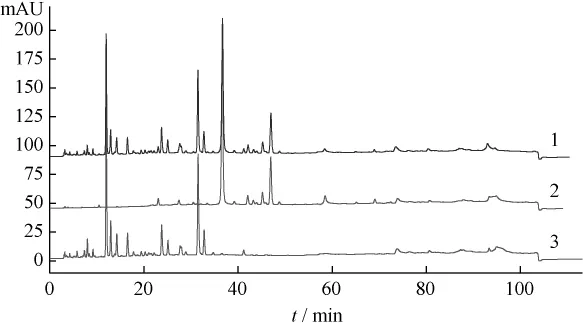
圖4 葛根芩連湯、黃芩藥材及陰性樣品HPLC圖
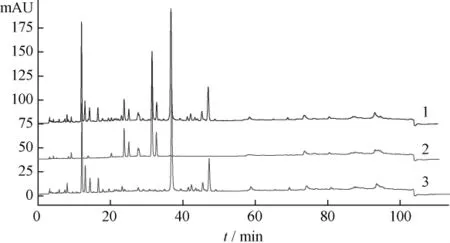
圖5 葛根芩連湯、黃連藥材及陰性樣品HPLC圖
2.5 結果 在葛根芩連湯指紋譜中均能找到葛根、黃芩和黃連藥材的特征峰群,其中 α 值為0.482、0.612、0.664、1.000、1.080、1.187、1.375、1.478、1.617、2.397 和 3.404(以葛根素為內參照峰)為葛根藥材的特征峰群;α值為0.633、0.753、1.000、1.070、1.147、1.179、1.232、1.279、1.595、1.884和1.998(以黃芩苷為內參照峰)為黃芩藥材的特征峰群;α 值為 0.132、0.292、0.438、0.645、0.798、0.879、1.000、1.040、2.935和2.986(以鹽酸小檗堿為內參照峰)為黃連藥材的特征峰群。結果表明所建立的數字化色譜指紋譜中可找出用以鑒定各藥材的較全面的特征峰群,可實現對復方中中藥材的鑒定。
3 討論
葛根芩連湯中成分復雜,含有多種生物堿、黃酮類、皂苷類成分,方中的黃酮類和生物堿類物質分別進行反相高效液相色譜分析時所適用的流動相條件差別較大。本研究考察了甲醇-水,甲醇-0.5%甲酸,甲醇-水(含0.5%甲酸和0.5%三乙胺)以及乙腈-水(含0.5%甲酸和0.5%三乙胺)等流動相對樣品組分分離的影響,確定了最佳流動相配比。同時比較了在 210,220,250,270,280,346,365 nm 不同檢測波長下的圖譜,結果顯示在270nm波長下,峰數目較多,各峰高低比例適宜,且基線平穩。建立的色譜條件可實現弱酸性和弱堿性物質同時分析,且在很大程度上解決了弱酸性和弱堿性物質共存時,弱堿性物質峰形差的問題。
藥材來源的峰在復方中的表達可以劃分一定的指紋區域,其中5~20 min主要來自葛根,20~35 min主要來自黃連,35~70 min主要來自黃芩,直觀的指紋圖譜是評價、控制中成藥質量的有效方法,也是中藥研究的一種模式。但這樣的方法有一定誤差且無量化指標。目前由于對照品有限,不能較全面地對指紋圖譜中的主要色譜峰一一定性,本研究通過分析復方、待鑒藥材和陰性樣品的數字化色譜指紋譜,找到了葛根、黃芩和黃連的特征峰群,可用于復方中上述藥味的鑒別,這也為今后葛根芩連湯化學配伍研究奠定了基礎。
通過對所建立的數字化色譜指紋譜分析,發現在全方中保留時間為57.285、81.309、88.250 min為葛根、黃芩和黃連藥材及各自的陰性樣品中均未檢測到的;提示上述成分為復方形成過程中產生的新成分,為進一步深入研究上述問題,需在中醫理論指導下,設計不同配伍的藥味在確定色譜條件下分析,建立相應的數字化色譜指紋譜進行更為深入地研究,如能結合液-質聯用等技術,則有望搞清指紋譜中相關成分的結構,從而為闡明其藥效物質基礎提供科學依據。
本研究在確定的色譜條件下對炙甘草進行如上述藥材的鑒定,結果不理想,炙甘草藥材出峰非常少,在全方中只檢出了保留時間為20.896 min處的甘草苷,含量非常低,未檢測到甘草酸。筆者將炙甘草單獨用甲醇提取后在確定色譜條件分析,可檢出成分較在全方中多,分析原因可能是全方水煎后引起炙甘草中成分變化,有文獻報道葛根芩連湯中黃連可降低其中甘草酸含量[7]。
[1]李順保.傷寒論版本大全[M].北京:學苑出版社,2001.
[2]劉 斌,石任斌,朱麗君,等.苦參湯黃酮類成分HPLC指紋圖譜及其與組方藥味黃芩和苦參的相關性研究[J].中國中藥雜志,2007,32(16):1631.
[3]洪筱坤.中藥數字化色譜指紋譜[M].上海:上海科學技術出版社,2003.
[4]王新宏,安 睿,鄒 云,等.數字化色譜指紋譜技術在雙黃連制劑質量及藥材鑒定中的應用[J].中草藥,2002,33(2):115.
[5]安 睿,王新宏,周思麗,等.HPLC測定不同產地丹參藥材中脂溶性成分分析[J].中成藥,2004,26(4):249.
[6]安 睿,王新宏,唐 瑩,等.不同產地丹參藥材中水溶性成分分析[J].中成藥,2005,27(7):812.
[7]戴開金,羅佳波,吳昭暉,等.配伍對葛根芩連湯中甘草酸含量的影響[J].中草藥,2003,34(12):1084.