濃縮六味地黃丸中馬錢苷的藥代動力學研究
2010-01-30 06:23:18肖子曾李興豐廖建萍
中成藥 2010年10期
關鍵詞:血清
戴 冰, 肖子曾, 梅 君, 冷 旺, 李興豐, 趙 婧, 廖建萍, 彭 柳
(1.湖南中醫藥大學第一附屬醫院,湖南長沙410007;2.湖南中醫藥大學,湖南長沙410007)
六味地黃丸為棕褐色的濃縮丸,味微甜、酸、略苦,它是由六味中藥材組成:熟地黃、山茱萸、牡丹皮、山藥、茯苓、澤瀉。諸藥配伍,共奏滋陰補腎,用于腎陰虧損、頭暈耳鳴、腰膝酸軟、骨蒸潮熱 、盜汗遺精、消渴[1]。其有效成分主要有苷類,如芍藥苷、馬錢苷、地黃苷等;酚性成分,如丹皮酚等;萜類,如澤瀉醇 A、B、C等及一些有機酸如沒食子酸、熊果酸等[2]。其中馬錢苷為2010版《中國藥典》收載的六
味地黃丸指標成分之一,近年來對六味地黃丸中馬錢苷的研究較多[3-7],但大多集中在體外成分含量測定方面,其體內藥代動力學研究鮮有報道。本研究在參考體外研究的基礎上,采用反相高效液相色譜法測定大鼠血清中馬錢苷的濃度,研究大鼠灌胃給藥后的體內藥代動力學特點,這對臨床合理用藥及劑型的改進都將具有指導意義,同時為中藥復方的物質基礎研究也提供了新的思路。
1 儀器與試藥
1.1 儀器 Agilent-1100高效液相色譜儀(包括四元梯度泵、真空脫氣機、Chemstation software工作站,SPD-10AVP二極管陣列檢測器),DT-100電子分析天平(北京光學儀器廠),DL-360A超聲清洗機(上海之信儀器有限公司);MILLI-QB純凈水發生器(美國MILLIPORE公司)。
1.2 試藥 濃縮六味地黃丸(批號:20080115,九芝堂股份有限公司);戊巴比妥鈉(批號:86-01-22,上海化學試劑采購供應站分裝廠);馬錢苷標準品(河北醫科大學藥學院天然藥物化學教研室,白色粉末,純度>99.5%);乙腈(色譜純)、磷酸(分析純)、甲醇(色譜純)、乙醇(色譜純)、雙蒸水。
1.3 實驗動物 健康Wistar大鼠,清潔級,♀♂各半,體重(200±20)g,湖南農業大學實驗動物中心提供,合格證號:醫動字第20-002號。
2 方法與結果
2.1 色譜條件 Kromasil C18色譜柱(250 mm×4.6 mm,5 μm);Nova-Pak C18Guard-PakTM保護柱。流動相:0.05%磷酸水溶液(A)-乙腈(B);檢測波長:236 nm;柱溫:30℃;流速:1 mL/min;分析時間55 min,洗脫梯度見表1。
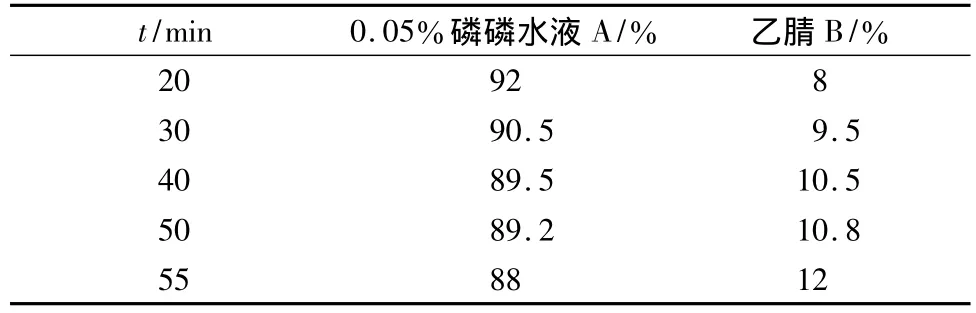
表1 梯度洗脫程序
2.2 供試品溶液的制備 取六味地黃丸400丸(每8丸相當于生藥材量3 g),加純凈水約300 mL,攪拌,煮沸,使其藥液濃縮至0.6 g/mL(按生藥材含量計),供實驗用。
2.3 對照品溶液的制備 取馬錢苷對照品適量,加空白大鼠血清溶解,馬錢苷濃度為210 μg/mL,臨用前用空白血清稀釋至各所需濃度。
2.4 血清樣品的制備 大鼠按20 mL/kg給予供試品灌胃后,用1%戊巴比妥鈉腹腔注射麻醉,經肝門靜脈取血5 mL,靜置4 h,3 000 r/min離心15 min,取上清液1 mL,加入0.2 mL 70%高氯酸,快速搖勻,3 000 r/min條件下離心15 min,取上清液過0.45 μm微孔濾膜,備HPLC分析,血清-20℃冷凍保存,供實驗用[8]
2.5 藥動學試驗 取Wistar大鼠,清潔級,♀♂各半,體重(200±20)g。按20 mL/kg給予供試品灌胃,給藥后分別于不同時間(0、5、10、15、30、45、60、120、180、240、360、450 min)肝門靜脈采血,每個時間點6只大鼠。取血后按2.4項下方法制備含藥血清,進樣20 μL,測量色譜峰面積,計算血藥濃度。
2.6 色譜系統適應性試驗
2.6.1 方法確證 在上述色譜條件下,馬錢苷的分離效果好,空白血清中的內源性物質及其代謝產物,含藥血清中的其他物質成分均不干擾測定。理論塔板數不小于4 000,保留時間約為42.5 min。空白血清,空白血清中加入馬錢苷對照品,含藥血清及含藥血清加入馬錢苷對照品色譜圖,見圖1。
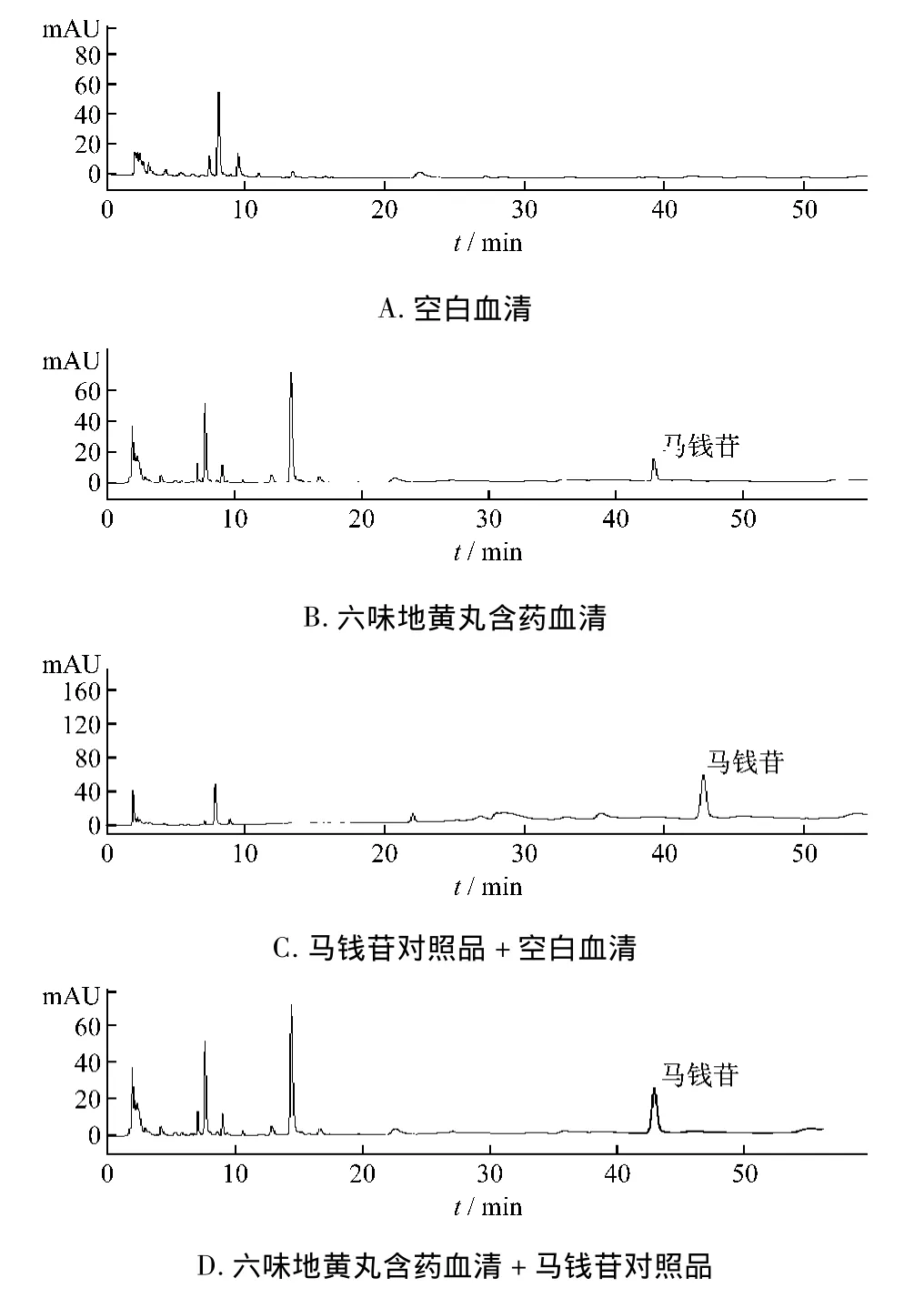
圖1 空白血清、對照品、含藥血清HPLC圖
2.6.2 標準曲線繪制 對照品溶液用空白血清稀釋成濃度分別為 2.6、5.2、10.5、21、42 μg/mL 的溶液,進樣10 μL,以峰面積積分值為縱坐標,進樣量(ng)為橫坐標做標準曲線,求得回歸方程,馬錢苷回歸方程為:Y=1.092 1X+27.064,r=0.999 1,線性范圍:26~420 ng。
2.6.3 精密度試驗 取同一濃度的對照品溶液,重復進樣5次,馬錢苷平均峰面積RSD為1.27%,結果表明:儀器精密度良好,符合體內藥物濃度測定的要求。
2.6.4 回收率試驗 按2.3項下方法制備不同濃度含藥血清標準液,經測定,不同濃度下均有高而穩定的回收率,各濃度回收率符合體內藥物濃度測定的要求,RSD為4.01%,結果見表2。

表2 回收率試驗結果
2.6.5 穩定性試驗 取空白血清,加入適量標準品,配置成終濃度為55.0 μg/mL馬錢苷血清樣品,分別于 2、4、6、8、24 h 分別進樣分析 1 次,連續測定5次,計算RSD(變異系數)值,RSD=0.55%,表明:血清樣品24 h內穩定性良好。
2.7 藥動學參數 大鼠灌胃后,分別于不同時間點測定馬錢苷的血藥濃度。采用3P97藥動學軟件進行自動擬合處理。以理論血藥濃度值與實際測定值的相關系數最大和AIC最小作為判斷標準,權重1/C。結果馬錢苷的藥動學曲線經擬合符合一室模型,主要藥動學參數見表3,平均血藥濃度-時間曲線見圖2。

表3 灌胃給藥后馬錢苷的主要藥動學參數
3 討論
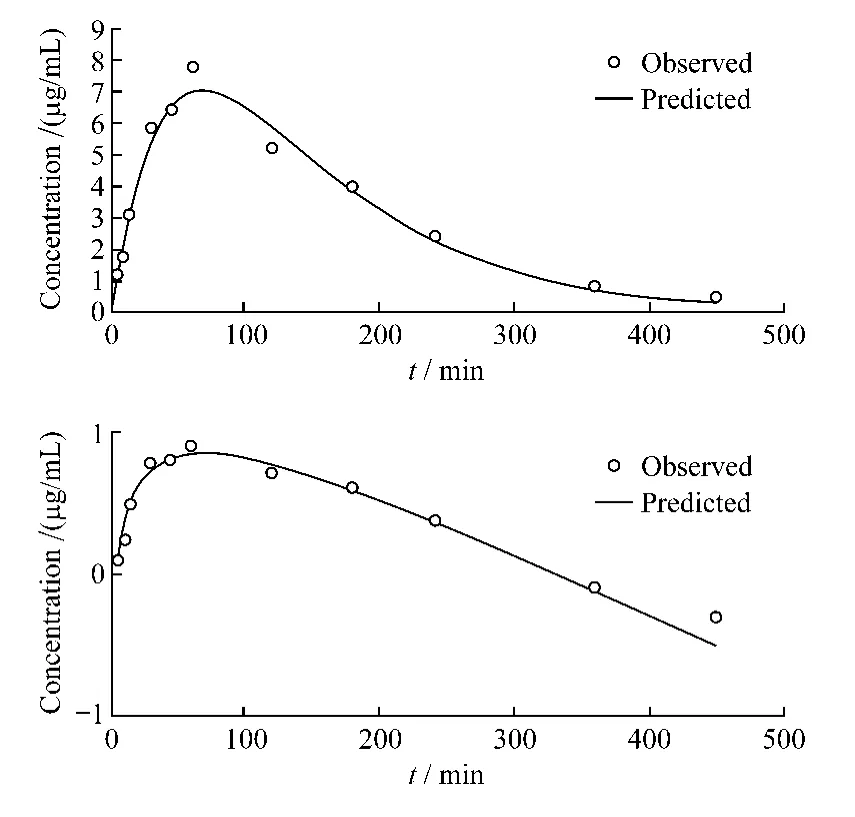
圖2 平均血藥濃度-時間曲線(n=6,±s)
3.1 馬錢苷為2010版《中國藥典》收載的六味地黃丸指標成分之一,近年來對六味地黃丸中馬錢苷的研究較多[3-7],但大多集中在體外成分含量測定方面,其體內藥代動力學研究鮮有報道。本研究在體外研究的基礎上,采用反相高效液相色譜法測定大鼠血清中馬錢苷的濃度,研究大鼠灌胃給藥后的體內藥代動力學特點,這對臨床合理用藥及劑型的改進都將具有一定的指導意義。
3.2 本研究以血藥濃度法作為手段,采用高效液相色譜技術,在參考有關文獻[7]的基礎上建立了適合本研究的色譜條件和血清樣品制備方法。通過比較空白血清、空白血清加馬錢苷對照品、六味地黃丸含藥血清以及六味地黃丸含藥血清加馬錢苷對照品的色譜圖,可很好地對六味地黃丸含藥血清中的馬錢苷進行定性分析;且能將大鼠血清中目標成分與血清中蛋白雜峰、內源性物質、六味地黃丸的其他成分以及這些成分的體內代謝產物和結合性成分很好分離。此方法重現性好,操作程序簡便,為相關制劑中馬錢苷的入血分析提供參考。
3.3 檢測波長的選擇 本實驗采用二極管陣列檢測器,在分析時進行200~400 nm紫外掃描,確定馬錢苷在236 nm有最大吸收,故選用236 nm作為檢測波長。
3.4 色譜條件的優化 實驗中考察了不同系統的流動相進行梯度洗脫篩選試驗,包括:①乙腈-水溶液②甲醇-水溶液③乙腈-0.05%磷酸水溶液④甲醇-0.05%磷酸水溶液多個梯度洗脫系統作為流動相進行考察。結果表明:乙腈-0.05%磷酸水溶液梯度洗脫系統最佳,色譜峰峰型好,各色譜峰分離度高,保留時間適宜,故選用該流動相系統作為六味地黃丸含藥血清含量測定的洗脫系統。
3.5 由于中藥復方所含成分的復雜性,口服后有效成分含量偏低,經制劑處理,服用時胃腸道的選擇性吸收及肝臟的首過效應等原因,其在血液中的含量大大減少,經HPLC分析時難以檢出,故本研究采用肝門靜脈采血法,因為經肝門靜脈采血具有原型入血成分血藥濃度較高(藥物經消化道吸收,尚未被肝臟代謝)、操作簡便、清潔衛生等優點,從而確保目標成分的檢出。
3.6 由于考慮到血清沉淀蛋白及過濾進樣消耗量大,所以選用大鼠單次采血,這樣能保證分析的正常進行,但缺點是會造成個體差異誤差較大,本實驗選用多個樣本重復求平均值的方法減小誤差。
[1]李洪珍.六味地黃丸含量測定方法的探索[J].菏澤醫學專科學校學報,2007,19(2):42-43.
[2]曾常青,曾 宇.六味地黃湯中4種有效成分的含量測定[J].中國醫院藥學雜志,2007,27(8):1034-1035.
[3]閻東海.HPLC測定濃縮六味地黃丸中馬錢苷含量[J].中國現代中藥,2007,9(7):19,23.
[4]馮 堃.HPLC法測定濃縮六味地黃丸中馬錢苷含量[J].鄭州大學學報,2008,43(4):790-791.
[5]譚秋紅.RP-HPLC法測定六味地黃丸中馬錢苷的含量[J].中國醫學工程,2007,15(7):601-602.
[6]唐益華,肖凱華.RP-HPL C法測定濃縮六味地黃丸中馬錢苷的含量[J]. 中國藥事,2007,21(10):834-835.
[7]謝躍生,張振清.六味地黃湯中馬錢子素小鼠體內藥代動力學研究[J].中草藥,2002,33(9):548-550.
[8]王喜軍,張 寧.六味地黃丸的血清藥物化學研究[J].中國天然藥物,2004,2(4):219-222.
猜你喜歡
中老年保健(2021年3期)2021-08-22 06:50:04
天津醫科大學學報(2021年2期)2021-03-29 05:31:08
昆明醫科大學學報(2021年1期)2021-02-07 01:06:36
現代臨床醫學(2021年1期)2021-01-26 00:56:02
昆明醫科大學學報(2020年12期)2021-01-26 00:44:04
中華養生保健(2020年4期)2020-11-16 01:31:40
中西醫結合肝病雜志(2020年2期)2020-10-27 02:18:50
豬業科學(2018年8期)2018-09-28 01:27:38
中成藥(2017年8期)2017-11-22 03:18:47
川北醫學院學報(2015年5期)2015-12-05 08:22:29