腹主動脈縮窄大鼠模型血漿血管緊張素Ⅱ的改變
2010-06-07 04:58:24趙凌杰焦東東張永文商瑋蔡輝
河北醫藥 2010年3期
趙凌杰 焦東東 張永文 商瑋 蔡輝
心肌纖維化(myocardial fibrosis,MF)是心室重塑的重要表現,是各種心血管疾病共有的進程,嚴重影響患者心功能甚至生活質量,腎素-血管緊張素系統(RAS)過度激活在其中發揮重要作用。血管緊張素Ⅱ(AngⅡ)作為誘導MF、心肌細胞肥大過程中的主要介質,對細胞間質膠原的沉積至關重要,其過度表達導致心肌代謝及功能異常,使其僵硬度增高,舒縮功能障礙。我們利用腹主動脈縮窄法誘導大鼠后壓力超負荷所致MF模型,探討其血漿中AngⅡ表達的改變。
1 材料與方法
1.1 實驗動物與試劑 雄性SD大鼠30只,體重180~200 g,由南京軍區南京總醫院實驗動物中心供應。飼養環境(22±2)℃,常規飼料喂養。即用型血管緊張素Ⅱ放免試劑盒購自北京海瑞生物工程公司,放免測定在南京軍區南京總醫院放免中心完成。
1.2 分組及模型制備 SD大鼠隨機取14只作為假手術組,余為模型組。模型組根據Doering等的方法,參照其腹主動脈縮窄法,用3%戊巴比妥鈉40mg/kg腹腔注射麻醉,固定,剃毛,常規消毒,分層打開腹腔,在左腎上緣分開后腹膜等軟組織,暴露腹主動脈并在雙腎動脈上方分離腹主動脈,用銀夾造成腹主動脈部分狹窄(銀夾內徑0.7mm)分層關閉腹腔。假手術組只打開腹腔、分離腹主動脈,不用銀夾縮窄。
1.3 觀測指標
1.3.1 左心室質量指數(LVMI):左心室與室間隔的重量(濕重)為左心室重量,用上海精科天平廠生產的JA1003型電子天平稱重。左心室重量(mg)/體重(g)為LVMI(‰)。
1.3.2 左心室心肌病理形態HE染色:左心室心肌常規石臘切片采用HE染色,經過脫臘、染色、脫水、二甲苯透明及中性樹膠封片等步驟,在光鏡下觀察左心室心肌組織病理形態改變。
1.3.3 AngⅡ放免測定:腹主動脈取血3 ml,迅速注入冰水冷卻的抗凝管(10%EDTA二鈉)中搖勻,即刻再放回冰水浴中冷卻,4℃ 1000 r/min,離心5 min,分離血漿。血漿 -70℃保存。測定時冰凍標本在流動的冷水浴中快速融化,在冰水浴中,加樣操作后,3500 r/min,離心15 min,吸棄上清,測定各管沉淀的放射性(cpm)。
1.4 統計學分析應用SPSS 11.5統計軟件,計量資料以表示,采用t檢驗,P<0.05為差異有統計學意義。
2 結果
2.1 造模后模型組死亡6只,8周實驗結束剩10只,死亡率為37.5%(6/16)。假手術組死亡2只,8周實驗結束剩12只;8周死亡率14.3%(2/14)。
2.2 LVMI 腹主動脈縮窄大鼠造模8周(模型組)時LVMI為(2.91±0.32)‰較假手術組(2.10±0.12)‰顯著升高(P<0.01)。
2.3 左心室心肌病理形態HE染色 光鏡下可見假手術組造模8周時左心室心同HE染色改變基本一致,心肌纖維排列整齊,心肌細胞大小、形態正常。模型組造模8周左心室心肌病理形態HE染色顯示,心肌纖維排列紊亂,細胞橫徑增大。見圖 1、2。

圖1 假手術組左心室心肌造模8周時(HE×100)
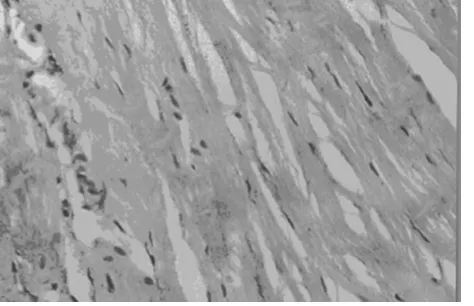
圖2 模型組左心室心肌造模8周時(HE×100)
2.4 血漿AngⅡ濃度的改變 造模8周時模型組血漿AngⅡ濃度為(640±220)pg/ml,較假手術組(419±76)pg/ml顯著升高(P <0.01)。
3 討論
AngⅡ是RAS系統的重要的生物活性肽,在多種心血管疾病發生發展中起重要作用。病理條件下,多種因素可誘導腎素釋放增加,作用于血管緊張素原,使其生成10肽化合物AngⅠ;AngⅠ可在多種酶作用下轉化為AngⅡ。傳統多認為ACE途徑是AngⅠ轉化為AngⅡ的主要途徑。糜蛋白酶是一種儲存的惰性糜蛋白酶樣的絲氨酸蛋白酶,Bacani等[1]認為糜蛋白酶可不依賴血管緊張素轉換酶(ACE)的合成AngⅡ,這一途徑脈管損傷后糜蛋白酶釋放入間質立即啟動,并且糜蛋白酶在間質組織中AngⅠ轉化為AngⅡ方面占主要優勢。糜蛋白酶尚可促進內皮素1前體的成熟[2],進而影響心肌間質改變。此外,AngⅠ轉化為AngⅡ也可經胰蛋白酶、激肽釋放酶、組織蛋白酶G的作用途徑完成,這一觀點解釋了臨床血管緊張素轉換酶抑制劑應用過程中出現的逃逸現象。
AngⅡ受體有AT1受體和AT2受體,AngⅡ的生理作用幾乎均由AT1受體介導,如血管收縮、醛固酮分泌、兒茶酚胺釋放、水鈉潴留、細胞增殖等。多數試驗證實AT1受體抑制劑ARB可通過阻斷 RAS系統和其他旁路途徑生成的AngⅡ與AT1受體結合,阻斷導致MF的途徑,同時增加為AngⅡ與 AT2的結合,產生擴張血管,刺激一氧化氮和前列環素的增加,并防止心肌及血管壁肥厚的作用,從而延緩心力衰竭的進程。厄貝沙坦通過阻滯AngⅡ受體,減少醛固酮分泌和交感神經的活性,改善心功能、能量代謝及毛細血管再生,抑制膠原的形成和改善MF[3]。但Varagic等[4]研究顯示ARB可改善鹽相關心腎的功能和結構異常而不降低血壓,實驗數據顯示AngⅡ(通過AT1受體)可能部分參與鹽負荷獨立壓力作用所致高血壓靶器官損害。AT2受體分布較AT1廣泛但目前對其功能了解不完全,大多認為可對抗AT1受體效應,如擴張血管、抑制生長、產生凋亡等。Jiang等[5]研究顯示AT2受體可通過調節Ⅰ型膠原和金屬蛋白酶組織抑制劑-1(TIMP1)mRNA水平影響心肌成纖維細胞的膠原代謝,從而發揮其逆轉MF作用。自20世紀80年代起,人們發現除循環中的RAS外,在許多組織如心臟、血管、腦、腎等組織中也發現有腎素、Ang、ACE mRNA的表達,提示尚存在局部組織的RAS。雖然局部組織RAS各組分既可從血漿中攝取,也可在局部產生,但是局部組織的RAS生成及清除AngⅡ時,其酶學特征(底物濃度、動力學)與血漿中RAS不同。Yamamoto等[6]研究顯示ACE2的缺失能促進壓力超負荷誘導的心臟功能障礙,這一過程通過增加局部AngⅡ完成,說明心血管組織中的RAS在高血壓、心血管重塑等的發生發展過程中起重要作用。另外,新提出的腦RAS概念包括ACE2、AngⅠ~Ⅶ、腎素原和mas受體,RAS轉基因鼠腎素和血管緊張素原的過復制顯示腦RAS的重要性,腦中腦腎素原生成的AngⅡ可發揮神經調節作用,可調控交感神經活性和壓力反射[7],進而影響心臟負荷甚至心肌重塑的進程。
MF是心室重塑的重要表現,是各種心血管疾病共有的進程,研究MF模型具有重要的意義。MF作為一種病理學改變,存在于高血壓及心力衰竭等慢性心血管疾病模型進程中。本研究顯示,造模8周時模型組左心室質量指數較假手術組顯著升高(P<0.01);左心室心肌病理形態HE染色心肌纖維排列紊亂,細胞橫徑增大,較假手術組變化明顯,符合心肌纖維化細胞外基質改變。近來發現,CCN家族與心血管疾病有密切關系,現在普遍接受的是CCN蛋白并非生長因子,而是其他分子的修飾蛋白,尤其與細胞外基質代謝密切相關[8]。CCN蛋白參與有絲分裂、吸附、細胞程序性凋亡及細胞外基質生成、增長、停止,甚至多種細胞型的遷移。心肌細胞、成纖維細胞及細胞外基質均表達CCN1,而CCN1表達又受機械應力、缺血、缺氧刺激以及神經體液因子如AngⅡ與腎上腺素等誘導調節。目前CCN家族被認為是多種疾病新的有效治療靶點,具有重要的臨床研究意義。
本研究顯示造模8周時模型組血漿AngⅡ水平較假手術組顯著升高(P<0.01),提示大鼠腹主動脈縮窄所致心臟后負荷的高壓力可能通過提高血漿AngⅡ水平而發揮促進MF的作用。這一過程中,心肌細胞和基質結構發生重塑,心肌僵硬度增高,左心室心肌中AT1受體蛋白表達水平發生改變,進一步提高AngⅡ的敏感性。AngⅡ在其中發揮重要作用,可通過TGF/Smad、NF-κB、cAMP、cGMP 及 CCN 等途徑促進核內膠原mRNA表達,導致心肌中膠原沉積及比例改變,最終導致MF。因此,探討AngⅡ及相關因子參與MF機制,對深入認識AngⅡ在心肌細胞外基質改變及心血管系統基質信號網絡的調控中的作用具有重要意義。
1 Bacani C,Frishman WH.Chymase:a new pharmacologic target in cardiovascular disease.Cardiol Rev,2006,14:187-193.
2 D’Orleans Juste P,Houde M,Rae GA,et al.Endothelin-1(1-31):from chymase-dependent synthesis to cardiovascular pathologies.Vascul Pharmacol,2008,49:51-62.
3 黃嵐.厄貝沙坦對老年高血壓左心室肥厚的作用.醫學研究生學報,2007,20:218-219.
4 Varagic J,Frohlich ED,Susic D,et al.AT1 receptor antagonism attenuates target organ effects of salt excess in SHRs without affecting pressure.Am J Physiol Heart Circ Physiol,2008,294:H853-858.
5 Jiang XY,Gao GD,Du XJ,et al.The signalling of AT2 and the influence on the collagen metabolism of AT2 receptor in adult rat cardiac fibroblasts.Acta Cardiol,2007,62:429-438.
6 Yamamoto K,Ohishi M,Katsuya T,et al.Deletion of angiotensin converting enzyme 2 accelerates pressure overload induced cardiac dysfunction by increasing local angiotensin II.Hypertension,2006,47:718-726.
7 Phillips MI,de Oliveira EM.Brain renin angiotensin in disease.J Mol Med,2008,86:715-722.
8 Yeger H,Perbal B.The CCN family of genes:a perspective on CCN biology and therapeutic potential.J Cell Commun Signal,2007,1:159-164.
猜你喜歡
童話王國·奇妙邏輯推理(2024年5期)2024-06-19 16:03:38
現代臨床醫學(2022年4期)2022-09-29 07:38:00
昆明醫科大學學報(2021年4期)2021-07-23 01:21:50
中學生數理化·七年級數學人教版(2020年10期)2020-11-26 08:24:50
河北畫報(2020年10期)2020-11-26 07:20:50
數學物理學報(2020年2期)2020-06-02 11:29:24
光學精密工程(2016年6期)2016-11-07 09:07:19
海南醫學(2016年8期)2016-06-08 05:43:00
中國衛生標準管理(2015年3期)2016-01-14 03:41:47
醫學研究雜志(2015年9期)2015-07-01 17:28:15
