
3-吡咯烷基苯并蒽酮的電子結(jié)構(gòu)和光譜性質(zhì)
2010-11-06 07:01:16何榮幸
物理化學(xué)學(xué)報 2010年9期
陳 奔 何榮幸 李 明
(西南大學(xué)化學(xué)化工學(xué)院,重慶 400715)
3-吡咯烷基苯并蒽酮的電子結(jié)構(gòu)和光譜性質(zhì)
陳 奔 何榮幸 李 明*
(西南大學(xué)化學(xué)化工學(xué)院,重慶 400715)
苯并蒽酮衍生物在新型熒光材料、非線性光學(xué)材料和液晶顯示材料等領(lǐng)域有較大的應(yīng)用前景.本文采用量子化學(xué)方法優(yōu)化了3-吡咯烷基苯并蒽酮的基態(tài)幾何結(jié)構(gòu)和第一單重激發(fā)態(tài)的幾何結(jié)構(gòu),并與X射線晶體衍射實驗值進行了對比.利用含時密度泛函理論(TD-DFT)的不同泛函,計算了3-吡咯烷基苯并蒽酮在氣相和溶劑中的吸收和發(fā)射光譜,考察了它的電子結(jié)構(gòu)和光譜特征,并分析了不同泛函、基組以及溶劑效應(yīng)對吸收和發(fā)射光譜的影響.計算結(jié)果表明:3-吡咯烷基苯并蒽酮的最強吸收和發(fā)射光譜都是具有π→π*躍遷特征的電荷轉(zhuǎn)移(CT)態(tài);泛函B3LYP能較好地重現(xiàn)實驗吸收能;而對于具有分子內(nèi)電荷轉(zhuǎn)移特征的激發(fā)態(tài),泛函MPWK能較好地重現(xiàn)實驗發(fā)射能.溶劑效應(yīng)的計算表明,不同極性的溶劑對3-吡咯烷基苯并蒽酮的吸收光譜和發(fā)射光譜的影響較小.理論預(yù)測的光譜與實驗結(jié)果一致.
密度泛函理論; 吸收和發(fā)射光譜; 激發(fā)態(tài); 分子內(nèi)電荷轉(zhuǎn)移; 溶劑效應(yīng)
有機化合物的分子內(nèi)電荷轉(zhuǎn)移(ICT)是許多材料科學(xué)、化學(xué)、物理學(xué)和生物過程中最基本的過程,研究表明這些化合物一般都包含有電子供體(D)和電子受體(A)[1-5].若電子供體和電子受體之間有高度離域的π結(jié)構(gòu)相連,則這種化合物很可能作為光敏材料在分子電子學(xué)、非線性光學(xué)(NLO)材料、太陽能電池、生物化學(xué)、熒光探針等領(lǐng)域有著潛在的應(yīng)用前景[6-8].
苯并蒽酮衍生物由于具有高效熒光性質(zhì)、高耐熱和耐光性,在過去幾十年已經(jīng)被廣泛地應(yīng)用于紡織品和聚合物的分散染色劑、熒光涂料和激光染料[9-10].近年來的一些研究表明,它的3-氮取代衍生物具有特殊的光學(xué)性質(zhì),有望應(yīng)用于新型熒光材料、非線性光學(xué)材料和液晶顯示材料等領(lǐng)域,因此很多科學(xué)家對此進行了廣泛研究[11-14].最近,Kirilova等[15]合成了一種新型的激發(fā)態(tài)分子內(nèi)電荷轉(zhuǎn)移化合物—3-吡咯烷基苯并蒽酮,并應(yīng)用X射線晶體衍射實驗表征了它的結(jié)構(gòu),研究了它的吸收和發(fā)射光譜性質(zhì),根據(jù)它的熒光譜帶定性指出3-吡咯烷基苯并蒽酮存在強烈的激發(fā)態(tài)分子內(nèi)電荷轉(zhuǎn)移(CT)現(xiàn)象. Nepra"等[16]通過PPP-MO方法研究了苯并蒽酮衍生物的激發(fā)能和第一吸收光譜帶,并分析了基態(tài)和激發(fā)態(tài)電子密度分布與光譜性質(zhì)的聯(lián)系.Kosower等[17]的理論研究認為,苯并蒽酮衍生物的激發(fā)態(tài)是由一個平面的CT態(tài)衰減而來,并解釋了極性溶劑中熒光量子產(chǎn)率急劇減少的原因.作為一種重要的激發(fā)態(tài)分子內(nèi)電荷轉(zhuǎn)移化合物,研究3-吡咯烷基苯并蒽酮基態(tài)和激發(fā)態(tài)的電子結(jié)構(gòu)、吸收和發(fā)射光譜的性質(zhì)、溶劑極性對光譜的影響等具有重要的理論和實際意義.
本文選取圖1所示的3-吡咯烷基苯并蒽酮作為研究對象,采用含時密度泛函理論(TD-DFT)(所用泛函為B3LYP,B3P86,MPWK,BHandHLYP)和只考慮單激發(fā)的組態(tài)相互作用(CIS)方法,研究它的電子結(jié)構(gòu)和光譜性質(zhì),分析不同泛函和基組對吸收和發(fā)射光譜的影響,并討論溶劑效應(yīng)的影響.

圖1 3-吡咯烷基苯并蒽酮的結(jié)構(gòu)及原子編號Fig.1 Structure and atomic number of 3-pyrrolidinobenzanthrone
1 計算方法
采用密度泛函理論(DFT)[18-19]的 B3LYP[20-21]、B3P86[21-22]和MPWK[23-24]三個泛函和HF方法,在6-311++g**基組水平上對分子的基態(tài)幾何結(jié)構(gòu)進行了全優(yōu)化;第一單重激發(fā)態(tài)的幾何結(jié)構(gòu)采用CIS方法在相同基組水平進行了優(yōu)化,并通過頻率分析確認了其優(yōu)化結(jié)構(gòu)都處于勢能面的極小點.利用獲得的優(yōu)化結(jié)構(gòu),采用含時密度泛函理論的不同泛函計算了分子的吸收和發(fā)射光譜.對于溶劑效應(yīng),使用極化連續(xù)介質(zhì)模型(the polarzable continuum model, PCM)[25]來計算.全部計算均用Gaussian 03程序包[26]完成.
在TD-DFT的計算中,我們采用了B3LYP, B3P86,BHandHLYP[26]和MPWK四種泛函以及6-31+g*,6-311++g**兩個基組.之所以選擇這四個泛函是因為它們廣泛的適用性,特別是B3LYP幾乎變成了結(jié)構(gòu)優(yōu)化的一個標準泛函,而MPWK則是計算電子激發(fā)能常用的近似交換相關(guān)泛函之一.和B3LYP相比,MPWK雜化泛函對于很多激發(fā)態(tài),包括低的Rydberg態(tài)的計算結(jié)果和實驗值都吻合得更好[23-24,27].另外,用B3P86和BHandHLYP泛函計算了研究對象的吸收與發(fā)射光譜,考察不同泛函對本文研究對象的適用情況.
2 基態(tài)及激發(fā)態(tài)的幾何結(jié)構(gòu)
基態(tài)結(jié)構(gòu)的優(yōu)化采用B3LYP,B3P86,MPWK和HF方法在6-311++g**基組水平完成.表1給出了3-吡咯烷基苯并蒽酮在各種理論水平下,基態(tài)和激發(fā)態(tài)結(jié)構(gòu)參數(shù)與X射線晶體衍射實驗值的比較.結(jié)果顯示,基態(tài)苯并蒽酮環(huán)上的C—C鍵長在0.137-0.145 nm之間,介于標準C═C雙鍵(約0.134 nm)和C—C單鍵(0.153 nm)之間,顯示苯并蒽酮環(huán)上的電子分布是離域的.B3LYP/6-311++g**理論水平給出優(yōu)化結(jié)構(gòu)的二面角D(C15C16C12C13)=0.6° (0.2°,HF),D(C3C17C15C14)=178.3°(179.0°,HF),說明苯并蒽酮環(huán)有良好的共平面性,但由于C3位上的吡咯烷基團的取代,造成了與之相鄰的苯環(huán)共面性下降.泛函B3P86、MPWK也給出了相似的結(jié)果.
進一步比較基態(tài)和激發(fā)態(tài)的結(jié)構(gòu)參數(shù)可以看出,和基態(tài)的結(jié)構(gòu)參數(shù)相比,激發(fā)態(tài)的結(jié)構(gòu)參數(shù)發(fā)生了較大的變化.二面角D(C15C16C12C13)由基態(tài)時的0.2°(HF)變?yōu)榧ぐl(fā)態(tài)時的2.3°(CIS),二面角D(C3C17C15C14)由基態(tài)時的179.0°(HF)(178.3°, B3LYP)變?yōu)榧ぐl(fā)態(tài)時的177.6°(CIS),說明激發(fā)態(tài)時的苯并蒽酮環(huán)平面發(fā)生一定扭曲,共面性下降,電子離域程度減小.C7═O23鍵長由基態(tài)時的0.1226 nm(B3LYP)縮短為激發(fā)態(tài)時的0.1208 nm(CIS),說明激發(fā)態(tài)時苯并蒽酮環(huán)上羰基的電子云密度較基態(tài)時有所增加.基態(tài)時C3—N18鍵長為0.1411 nm(HF),介于標準C═N雙鍵(約0.136 nm)以及C—N單鍵(0.1475 nm)之間,二面角D(C19N18C3C22)=128.3° (130.1°,B3LYP),說明C3—N18有一定程度的離域,而激發(fā)態(tài)時C3—N18鍵長縮小為0.1375 nm,二面角D(C19N18C3C22)為137.8°,C3—N18鍵長縮小和氨基共面性的增加說明了激發(fā)態(tài)時的N18能更好地參與電子離域,后文討論中前線軌道的分析也表明,分子激發(fā)態(tài)時發(fā)生由N18到苯并蒽酮環(huán)上羰基的電荷轉(zhuǎn)移,使得N18原子帶有部分正電荷,因而導(dǎo)致了氨基平面性的增加,從而使得N18的π電子能更好地參與電子離域.

表1 3-吡咯烷基苯并蒽酮基態(tài)和激發(fā)態(tài)結(jié)構(gòu)參數(shù)與X射線晶體衍射實驗值Table 1 Calculated structural parameters for the ground and the first excited states of 3-pyrrolidinobenzanthrone and the experimental data
3 吸收光譜
3.1 氣相吸收光譜
在密度泛函方法B3LYP、B3P86和MPWK三個泛函以及HF方法優(yōu)化的基態(tài)構(gòu)型的基礎(chǔ)上,利用含時密度泛函理論計算3-吡咯烷基苯并蒽酮的吸收光譜.為了考察不同泛函對于計算結(jié)果的影響,采用B3LYP、B3P86和MPWK三個泛函分別在6-31+g*、6-311++g**兩個基組水平下,計算了3-吡咯烷基苯并蒽酮的氣相吸收光譜.TD-DFT能夠給出可靠的低激發(fā)態(tài)的性質(zhì)[28-29],但是對于較高激發(fā)態(tài)的結(jié)果誤差較大,所以我們計算了前10個激發(fā)態(tài)(表2中僅列出了諧振強度最大的前兩個).
從表2中發(fā)現(xiàn),不同理論水平優(yōu)化的基態(tài)構(gòu)型在相同TD-DFT泛函B3LYP下給出的理論結(jié)果基本一致,其諧振強度最大的躍遷態(tài)均為S1態(tài),相應(yīng)的垂直激發(fā)能分別為:2.74 eV(DFT-B3LYP優(yōu)化的幾何優(yōu)化結(jié)構(gòu)),2.75 eV(DFT-B3P86優(yōu)化的幾何結(jié)構(gòu)),2.75 eV(DFT-MPWK優(yōu)化的幾何結(jié)構(gòu))以及2.93 eV(HF優(yōu)化的幾何結(jié)構(gòu)).顯然HF方法優(yōu)化的基態(tài)幾何結(jié)構(gòu)計算的S1態(tài)的垂直躍遷能明顯比其他的高,原因在于HF方法沒有考慮電子的動態(tài)相關(guān),其優(yōu)化的基態(tài)結(jié)構(gòu)與真實值相差更大,這可以從表1中看到.
為了考察不同泛函在TD-DFT垂直激發(fā)能計算中的結(jié)果,我們采用了B3LYP,B3P86和MPWK三個泛函的平行計算.計算結(jié)果表明,B3LYP和B3P86兩個泛函給出的計算結(jié)果幾乎相同,而泛函MPWK給出了較低的垂直發(fā)射能.

表2 不同計算水平下3-吡咯烷基苯并蒽酮氣相吸收光譜的相關(guān)參數(shù)Table 2 Selected gas-phase absorption spectral parameters of 3-pyrrolidinobenzanthrone with different theoretical levels
為了考察基組對計算結(jié)果的影響,在TDB3LYP/6-31+g*和TD-B3LYP/6-311++g**兩個水平下對3-吡咯烷基苯并蒽酮S1態(tài)的垂直激發(fā)能進行了重復(fù)計算.分析表2中的計算結(jié)果,可以發(fā)現(xiàn)增大基組對于計算結(jié)果影響很小,這意味著使用中等大小的基組(6-31+g*)基本可以滿足本文所研究的體系.
為了分析激發(fā)態(tài)的電子結(jié)構(gòu),我們得到了不同理論計算水平下3-吡咯烷基苯并蒽酮的前線分子軌道.由于不同計算水平都給出了相近的前線分子軌道圖,這里只給出TD-B3LYP/6-311++g**理論水平下3-吡咯烷基苯并蒽酮的前線分子軌道,結(jié)果示于圖2.對于第一激發(fā)態(tài)S1,電子躍遷發(fā)生在最高占據(jù)分子軌道(HOMO)與最低空分子軌道(LUMO)之間(表2).如圖2所示,HOMO軌道是主要定域于N18原子上(還有部分在苯并蒽酮環(huán)平面的苯環(huán)上)的π型軌道,LUMO軌道則主要是定域于苯并蒽酮環(huán)平面蒽醌基團的π*型軌道,顯然S1態(tài)是一個具有π→π*特征的電荷轉(zhuǎn)移(CT)態(tài).這表明3-吡咯烷基苯并蒽酮在經(jīng)光激發(fā)之后,將直接生成電荷分離的激發(fā)態(tài),與實驗結(jié)果一致[15].第二激發(fā)態(tài)S2對應(yīng)于HOMO-1到LUMO的電子躍遷,HOMO-1軌道是位于羰基(C═O)上的n型軌道,因此S2態(tài)對應(yīng)的是n→π*的電子躍遷,計算的諧振強度f表明,S2態(tài)的諧振強度幾乎為0,這意味著S0→S2對應(yīng)的電子躍遷是禁阻的.

圖2 3-吡咯烷基苯并蒽酮的前線分子軌道圖Fig.2 Frontier molecular orbitals of 3-pyrrolidinobenzanthrone
3.2 液相吸收光譜
由于實驗[15]是在溶劑中進行的,因此為了更好地與實驗結(jié)果比較,需要考慮溶劑效應(yīng)對光譜的影響.實驗中所用溶劑分別是乙醇、氯仿和苯,相應(yīng)的介電常數(shù)分別為24.55、4.90和2.25.選取B3LYP/6-311++g**和B3P86/6-311++g**優(yōu)化的基態(tài)結(jié)構(gòu),利用泛函B3LYP、B3P86和MPWK,在6-311++g**和6-31+g*基組水平下,分別計算了3-吡咯烷基苯并蒽酮在苯、氯仿和乙醇中的吸收光譜,計算結(jié)果見表3.溶劑效應(yīng)采用PCM模型計算.
從表3可以看到,液相中諧振強度最大的激發(fā)態(tài)均為S1態(tài),對應(yīng)于HOMO→LUMO躍遷,與氣相結(jié)果一致.但是第二激發(fā)態(tài),S2的軌道躍遷順序發(fā)生了變化,它對應(yīng)于HOMO-2到LUMO的躍遷. HOMO-2軌道是定域于苯并蒽酮環(huán)平面苯環(huán)與N18原子上的π型軌道,因此在這種情況下S2態(tài)是一個π→π*的電子躍遷,與氣相結(jié)果的S3態(tài)一致,這是介質(zhì)的改變引起的.計算結(jié)果表明,S2態(tài)的諧振強度幾乎為0,這意味著S0→S2對應(yīng)的電子躍遷也幾乎是躍遷禁阻的.
考察溶劑效應(yīng)的計算結(jié)果,以TD-B3LYP/6-311++g**的計算結(jié)果為例,3-吡咯烷基苯并蒽酮在苯、氯仿和乙醇中的S1態(tài)對應(yīng)的垂直激發(fā)能分別為2.62、2.57和2.53 eV,而實驗給出的最大吸收波長分別為488 nm(2.54 eV)、495 nm(2.50 eV)和509 nm (2.46 eV),計算給出的躍遷能與實驗值[15]非常吻合,說明吸收光譜的計算是可信的.理論計算的結(jié)果表明,增大溶劑的極性,S1態(tài)對應(yīng)的垂直激發(fā)能和諧振強度的變化都很小,說明溶劑極性的改變對于吸收峰位置與電子結(jié)構(gòu)的影響很小,即溶劑效應(yīng)較小,這與實驗給出的苯→氯仿→乙醇的吸收光譜7-14 nm的輕微紅移,吸收光譜的溶劑效應(yīng)較小[15]相一致.泛函B3P86也給出相似的計算結(jié)果.另一方面,泛函MPWK給出的垂直激發(fā)能明顯偏低,在溶劑苯、氯仿和乙醇中,S1態(tài)對應(yīng)的垂直激發(fā)能分別為2.01、2.05和2.01 eV.
考察了幾何優(yōu)化構(gòu)型對垂直激發(fā)能的影響.同樣以3-吡咯烷基苯并蒽酮在苯中的吸收光譜為例,如表3所示,對于DFT-B3LYP和DFT-B3P86優(yōu)化的兩種不同的幾何構(gòu)型,TD-B3LYP/6-311++g**給出的S1態(tài)垂直躍遷能均為2.62 eV,這說明同屬于密度泛函方法優(yōu)化的幾何優(yōu)化構(gòu)型對于TD-DFT垂直躍遷能的計算結(jié)果沒有較大影響.分析極性更大的乙醇、氯仿的計算數(shù)據(jù)可以獲得相同的結(jié)論.
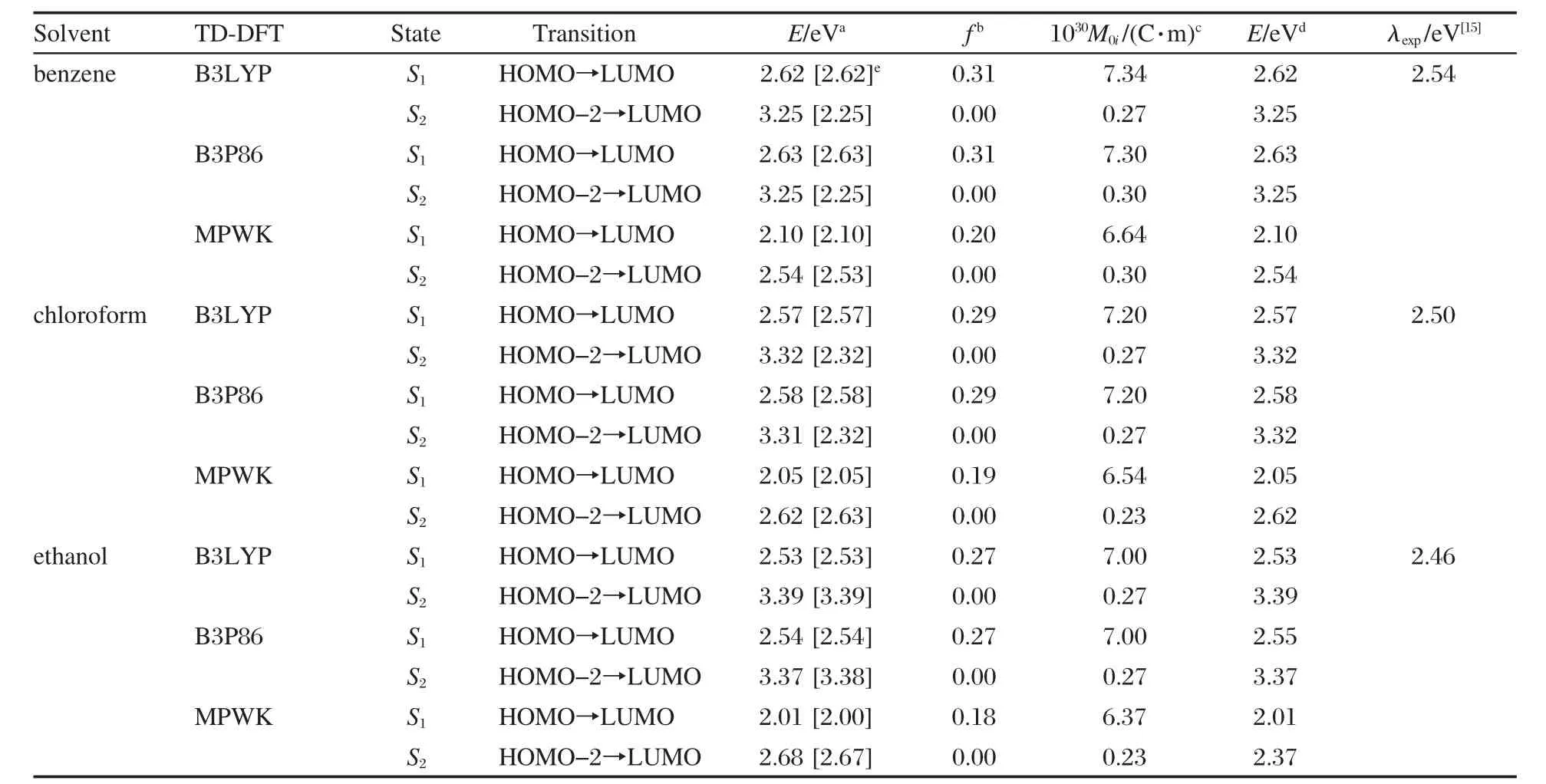
表3 不同計算水平下的3-吡咯烷基苯并蒽酮在乙醇、氯仿和苯中的吸收光譜參數(shù)Table 3 Selected absorption spectral parameters of 3-pyrrolidinobenzanthrone in ethanol,chloroform,and benzene with different calculated levels
從TD-B3LYP/6-31+g*與TD-B3LYP/6-311++g**的計算結(jié)果我們看到,基組大小的改變,并沒有對計算結(jié)果產(chǎn)生較大的影響,以3-吡咯烷基苯并蒽酮在苯中的吸收光譜計算為例,兩者給出的S1態(tài)對應(yīng)的垂直激發(fā)能幾乎一致.
為了更形象地比較不同溶劑中的吸收光譜計算結(jié)果,我們利用高斯函數(shù)和理論計算得到的各個激發(fā)態(tài)的躍遷能、諧振強度來模擬吸收光譜圖:
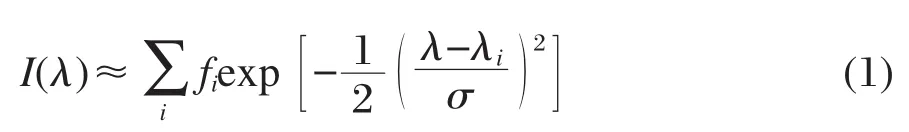
其中,i代表第i個激發(fā)態(tài);λi是計算得到的吸收波長(nm);fi指第i個激發(fā)態(tài)的計算諧振強度;λ和I(λ)分別為方程的自變量和因變量,其物理意義分別表示波長和振子強度;σ是以第i個激發(fā)態(tài)的吸收峰為中心的高斯半寬[28-29].將計算得到的三種溶劑中的激發(fā)態(tài)相應(yīng)物理量帶入公式(1)中,就得到如圖3所示的吸收光譜.很明顯,3-吡咯烷基苯并蒽酮在不同極性溶劑中的最強吸收峰位于470-500 nm之間,并且隨著溶劑極性的增強有著輕微的紅移.這與實驗結(jié)果[15]相一致.
4 發(fā)射光譜
4.1 氣相發(fā)射光譜
為了進一步研究激發(fā)態(tài)的性質(zhì)和發(fā)射光譜,在CIS/6-311++g**水平下優(yōu)化得到的激發(fā)態(tài)結(jié)構(gòu)的基礎(chǔ)上,利用含時密度泛函理論計算3-吡咯烷基苯并蒽酮的發(fā)射光譜.為了考察不同泛函對于計算結(jié)果的影響,我們采用B3LYP、B3P86、MPWK和 BHandHLYP四個泛函分別在6-31+g*和6-311++g**兩個基組水平下,計算了3-吡咯烷基苯并蒽酮的氣相垂直發(fā)射光譜,結(jié)果列于表4.
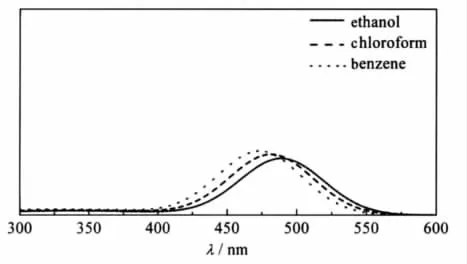
圖3 3-吡咯烷基苯并蒽酮在乙醇、氯仿和苯中的理論吸收光譜圖Fig.3 Calculated absorption spectra of 3-pyrrolidinobenzanthrone in ethanol, chloroform,and benzene
在TD-MPWK/6-311++g**水平下,3-吡咯烷基苯并蒽酮的垂直發(fā)射能約為2.13 eV,諧振強度為0.16,對應(yīng)的電子躍遷發(fā)生在LUMO與HOMO之間.從表4中發(fā)現(xiàn),不同泛函給出的理論垂直發(fā)射能相差較大.在6-311++g**基組水平下,泛函B3LYP和B3P86給出的S1態(tài)垂直發(fā)射能為2.45 eV,而泛函 BHandHLYP給出的垂直發(fā)射能為 2.84 eV. B3LYP和B3P86這兩個泛函給出的計算結(jié)果幾乎相同,和泛函BHandHLYP一樣,垂直發(fā)射能都明顯偏高,而泛函MPWK給出了與實驗值較符的結(jié)果.需要注意的是,在吸收光譜的計算中,泛函MPWK給出的計算結(jié)果與實驗值吻合得并不好,原因還有待進一步了解.
為了考察不同基組對于垂直發(fā)射能理論計算的影響,我們采用了6-31+g*基組重復(fù)上述計算.結(jié)果表明TD-MPWK/6-31+g*給出的氣相垂直發(fā)射能為2.16 eV,與TD-MPWK/6-311++g**的計算結(jié)果相比增大了0.03 eV,而后者的計算結(jié)果更為接近實驗值,說明在3-吡咯烷基苯并蒽酮的氣相垂直發(fā)射能的計算中加大基組對于理論值有一定改善.
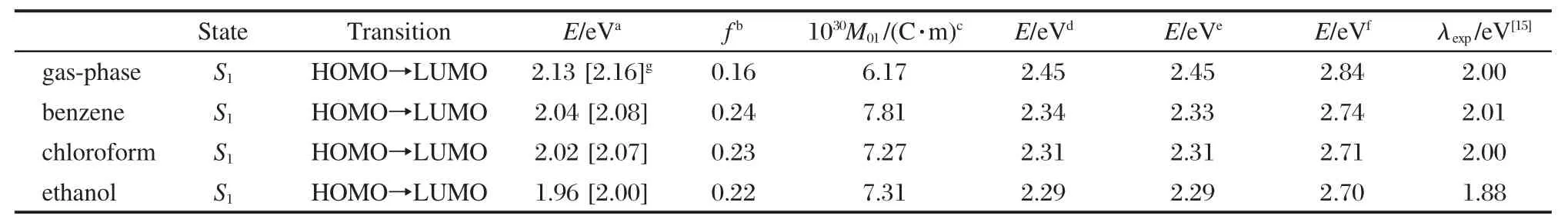
表4 不同計算水平下3-吡咯烷基苯并蒽酮在氣相及乙醇、氯仿和苯中的發(fā)射光譜參數(shù)Table 4 Selected emission spectral parameters of 3-pyrrolidinobenzanthrone in gas-phase,ethanol,chloroform, and benzene at different calculated levels
4.2 液相發(fā)射光譜
同吸收光譜一樣,發(fā)射光譜的溶劑效應(yīng)也需要考慮.在CIS/6-311++g**水平下優(yōu)化得到的第一單重激發(fā)態(tài)結(jié)構(gòu)的基礎(chǔ)上,利用含時密度泛函理論的B3LYP、B3P86、MPWK和BHandHLYP四個泛函,在6-311++g**基組水平下,采用PCM溶劑模型,計算了3-吡咯烷基苯并蒽酮在液相中的發(fā)射光譜,結(jié)果見表4.
從表4可以看到,液相中理論發(fā)射光譜的躍遷能級類型和能級順序與氣相中的完全相同,電子從LUMO軌道躍遷到HOMO軌道.與氣相發(fā)射光譜計算結(jié)果相似,在三種溶劑中,泛函MPWK給出的垂直發(fā)射能分別為2.04 eV(苯)、2.02 eV(氯仿)和1.96 eV(乙醇),與實驗值[15]吻合較好;泛函B3LYP, B3P86和BHandHLYP給出的垂直發(fā)射能計算值明顯偏高.
相比于氣相發(fā)射光譜,液相發(fā)射光譜發(fā)生了較大的紅移.在TD-MPWK/6-311++g**理論水平下, 3-吡咯烷基苯并蒽酮在苯、氯仿和乙醇中的垂直發(fā)射能相較于氣相垂直發(fā)射能都大約紅移了0.1 eV.在上述三種溶劑中,實驗[15]給出的最大發(fā)射波長分別為617 nm(2.01 eV)、627 nm(2.00 eV)和661 nm (1.88 eV),與計算值非常吻合,誤差僅為0.02-0.08 eV,說明發(fā)射光譜的計算是可信的.以TD-MPWK/ 6-311++g**的計算結(jié)果為例考察溶劑效應(yīng)的計算結(jié)果,我們發(fā)現(xiàn)3-吡咯烷基苯并蒽酮在苯、氯仿和乙醇中,對應(yīng)的垂直激發(fā)能均發(fā)生了大約0.08 eV的紅移(溶劑從苯→氯仿→乙醇),這與實驗[15]給出的溶劑效應(yīng)相一致.另外,對比溶劑相中的發(fā)射光譜和吸收光譜計算的計算結(jié)果,可以發(fā)現(xiàn)光譜發(fā)生了較大的Stocks位移,并且隨著溶劑極性的增加而增加(從苯→氯仿→乙醇分別為 133.25、133.09和 143.03 nm),這也與實驗結(jié)果[15]一致.
為了考察不同基組對于3-吡咯烷基苯并蒽酮液相中垂直發(fā)射能理論計算的影響,我們采用泛函MPWK在基組6-31+g*水平上做了重復(fù)計算,結(jié)果列于表4.計算結(jié)果表明增大基組對于發(fā)射光譜的理論值有所改善,但是并不明顯.

圖4 3-吡咯烷基苯并蒽酮的發(fā)射光譜Fig.4 Emission spectra of 3-pyrrolidinobenzanthrone(a)experimental emission spectrum(solid state)[15];(b)the calculated emission spectra in ethanol,chloroform,and benzene
采用與模擬吸收光譜圖一樣的公式,利用計算結(jié)果我們模擬了不同溶劑下的發(fā)射光譜圖,其結(jié)果與實驗光譜圖一起示于圖4.顯然,3-吡咯烷基苯并蒽酮在三種溶劑中的發(fā)射峰比較平坦,最強發(fā)射波長大約在600-640 nm之間,并隨著溶劑極性的增加而發(fā)生微小的紅移.分析圖4可以發(fā)現(xiàn),除了最強發(fā)射峰的位置稍微不同而外,理論模擬的發(fā)射光譜圖與實驗基本一致.
5 結(jié) 論
采用含時密度泛函理論(TD-DFT)的不同泛函以及不同基組,計算了3-吡咯烷基苯并蒽酮在氣相和溶劑相中的吸收和發(fā)射光譜,考察了它的電子結(jié)構(gòu)和光譜特征,并分析了不同泛函在計算垂直激發(fā)能和垂直發(fā)射能中的適用范圍以及不同基組對于計算結(jié)果的影響,討論了溶劑效應(yīng)對于吸收和發(fā)射光譜的影響.具體結(jié)論如下:
(1)不同理論水平下優(yōu)化的幾何構(gòu)型對于垂直躍遷能的影響較小,而不同泛函對于垂直躍遷能的計算值有很大影響.
(2)泛函B3LYP和B3P86能較好地重現(xiàn)實驗吸收能;而在發(fā)射光譜計算中,對于具有激發(fā)態(tài)分子內(nèi)電荷轉(zhuǎn)移特征的3-吡咯烷基苯并蒽酮,泛函MPWK給出的計算結(jié)果與實驗值更吻合.而增大基組對吸收光譜的計算影響較小,但對垂直發(fā)射能有一定改善.
(3)通過對前線分子軌道的分析,發(fā)現(xiàn)3-吡咯烷基苯并蒽酮的最強吸收和發(fā)射光譜都是具有π→π*躍遷特征的電荷轉(zhuǎn)移(CT)態(tài),這從理論上說明3-吡咯烷基苯并蒽酮在新型熒光材料、非線性光學(xué)材料等領(lǐng)域具有潛在的應(yīng)用價值.
(4)溶劑效應(yīng)對于吸收和發(fā)射光譜的影響較小, Stocks位移較小.理論預(yù)測的光譜與實驗結(jié)果一致.
1 Pourtois,G.;Beljonne,D.;Cornil,J.;Ratner,M.A.;Bredas,J.L. J.Am.Chem.Soc.,2002,124:4436
2 Zerza,G.;Sharber,M.C.;Brabec,C.J.;Sariciftci,N.S.;Gomez, R.;Segura,J.N.;Martin,N.;Srdanov,V.I.J.Phys.Chem.A, 2000,104:8315
3 Flouzat,C.;Besson,Y.;Mattio,A.;Bonnet,J.;Guillaumet,G. J.Med.Chem.,1993,36:497
4 Sobolewski,A.L.;Domecke,W.Chem.Phys.Lett.,1996,250: 428
5 Grabowski,Z.R.;Rotkiewicz,K.;Rettig,W.Chem.Rev.,2003, 103:3899
6 Bella,S.D.;Fragalá,I.L.;Ratner,M.A.;Marks,T.J.J.Am. Chem.Soc.,1993,115:682
7 Coe,B.J.;Harris,J.A.;Brunschwig,B.S.;Garin,J.;Orduna,J.; Coles,S.J.;Hursthouse,M.B.J.Am.Chem.Soc.,2004,126: 10418
8 Jiao,G.S.;Thoresen,L.H.;Burgess,K.J.Am.Chem.Soc.,2003, 125:14668
9 Krasovitskii,B.M.;Bolotin,B.M.Organic luminescent materials. New York:VCH Publishers,1988
10 Bunz,U.H.F.Chem.Rev.,2000,100:1605.
11 Hughes,G.;Bryce,M.R.J.Mater.Chem.,2005,15:94.
12 Kelley,T.W.;Baude,P.F.;Gerlach,C.;Ender,D.E.;Muyres,D.; Haase,M.A.;Vogel,D.E.;Theiss,S.D.Chem.Mater.,2004,16: 4413
13 Shirota,Y.;Kageyama,H.Chem.Rev.,2007,107:953
14 Logothetidis,S.Mater.Sci.Eng.B,2008,152:96
15 Kirilova,E.M.;Sergey,V.B.;Kirilov,G.K.;Kalnina,I.; Gerbreder,V.J.Lumin.,2009,129:1827
16 Nepra#,M.;Machalicky,O.;?eps,M.;Hrdina,R.;Kapusta,P.; Fidler,V.Dye and Pigments,1997,35:31
17 Kosower,E.M.;Dodiuk,H.J.Am.Chem.Soc.,1975,97:2167
18 Hohenberg,P.;Kohn,W.Phys.Rev.B,1964,136:864
19 Kohn,W.;Sham,L.J.Phys.Rev.A,1965,140:1133
20 Lee,C.;Yang,W.;Parr,R.G.Phys.Rev.B,1988,37:785
21 Becke,A.D.Phys.Rev.A,1988,38:3098
22 Perdew,J.P.Phys.Rev.B,1986,33:8822
23 Lynch,B.J.;Fast,P.L.;Harris,M.;Thruhlar,D.G.J.Phys.Chem. A,2000,104:4811
24 Lynch,B.J.;Zhao,Y.;Truhlar,D.G.J.Phys.Chem.A,2003, 107:1384
25 Cramer,C.J.;Truhlar,D.G.Chem.Rev.,1999,99:2161
26 Frisch,M.J.;Trucks,G.W.;Schlegel,H.B.;et al.Gaussian 03. Revision A.01.Pittsburgh,PA:Gaussian Inc.,2003
27 Wang,Y.L.;Wu,G.S.Acta Phys.-Chim.Sin.,2008,24:552 [王溢磊,吳國是.物理化學(xué)學(xué)報,2008,24:552]
28 He,R.X.;Duan,X.H.;Li,X.Y.J.Phys.Chem.A,2005,109: 4154
29 He,R.X.;Li,X.Y.Chem.Phys.,2007,332:325
Electronic Structures and Spectral Properties of 3-Pyrrolidinobenzanthrone
CHEN Ben HE Rong-Xing LI Ming*
(College of Chemistry and Chemical Engineering,Southwest University,Chongqing 400715,P.R.China)
Benzanthrone derivatives show great potential for use as new luminescent,nonlinear optical,and liquid crystalline materials.The geometries of the ground and the first excited states of 3-pyrrolidinobenzanthrone were optimized using quantum chemistry methods and the obtained structural parameters were compared with experimental data.The time-dependent density functional theory(TD-DFT)calculations were performed to estimate the absorption and emission spectra of 3-pyrrolidinobenzanthrone both in gas-phase and in solutions.In addition,the effects of different exchange correlation functionals,basis sets,and solvents on the absorption and emission spectra were analyzed in detail.We found that the strongest absorption and emission bands of 3-pyrrolidinobenzanthrone could be assigned to a charge transfer(CT)state with a π→π*character.The result of the B3LYP functional reproduces the experimental absorption spectrum very well and the MPWK functional accurately predicts the emission energy of the first excited state with an intramolecular charge transfer(ICT)feature.The calculated results indicate that solvent effects do not greatly influence the absorption and emission spectra.The theoretical results are in agreement with experimental observations.
Density functional theory; Absorption and emission spectrum; Excited state; Intramolecular charge transfer; Solvent effect
O641
Received:March 19,2010;Revised:April 23,2010;Published on Web:July 5,2010.
*Corresponding author.Email:liming@swu.edu.cn;Tel:+86-23-68253023.
The project was supported by the Natural Science Foundation of Chongqing,China(2009BB6002).
重慶市自然科學(xué)基金(2009BB6002)資助項目
?Editorial office of Acta Physico-Chimica Sinica
猜你喜歡
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
當(dāng)代陜西(2022年5期)2022-04-19 12:10:18
中學(xué)生數(shù)理化·中考版(2022年11期)2022-02-16 07:01:20
新世紀智能(數(shù)學(xué)備考)(2021年9期)2021-11-24 01:14:28
湘潮(上半月)(2021年4期)2021-07-20 08:05:28
汕頭大學(xué)學(xué)報(自然科學(xué)版)(2020年4期)2020-12-14 07:05:00
小哥白尼(趣味科學(xué))(2019年6期)2019-10-10 01:01:50
發(fā)明與創(chuàng)新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
學(xué)習(xí)月刊(2015年21期)2015-07-11 01:51:44
