
微乳液成核-離子交聯制備阿司匹林/殼聚糖納米微球及其體外釋放行為
2010-11-06 07:01:26金淑萍何文龑韓玉琦魏玉娟
物理化學學報 2010年9期
關鍵詞:殼聚糖
金淑萍 馮 雷 何文龑 韓玉琦 魏玉娟
(河西學院化學系,西部資源環境化學重點實驗室,甘肅張掖 734000)
微乳液成核-離子交聯制備阿司匹林/殼聚糖納米微球及其體外釋放行為
金淑萍 馮 雷*何文龑 韓玉琦 魏玉娟
(河西學院化學系,西部資源環境化學重點實驗室,甘肅張掖 734000)
以自制阿司匹林為藥物模型,殼聚糖(CS)為載體源,采用微乳液成核-離子交聯法制備了阿司匹林/殼聚糖納米緩釋微球.分別用傅里葉變換紅外(FTIR)光譜、場發射掃描電子顯微鏡(FESEM)、透射電子顯微鏡(TEM)、動態激光光散射(DLLS)、X射線粉末衍射(XRD)等表征了納米微粒的化學組成、外觀形貌、平均粒徑和粒徑分布、微球中殼聚糖的晶體結構以及阿司匹林的分布形態.結果表明,利用微乳液成核-離子交聯法制備的阿司匹林/殼聚糖微球平均粒徑約為88 nm且粒徑分布均勻,成核后殼聚糖結晶形態基本未變,阿司匹林以分子形態分布于微粒中,分子間未形成堆砌,為無定形態.采用UV-Vis分光光度計考察了微球的藥物包封率、載藥量,并對微球在生理鹽水和葡萄糖溶液中的釋藥行為進行跟蹤.結果表明,微球的載藥量可達55%,藥物包封率可達42%,實驗條件下具有較好的藥物緩釋作用.
殼聚糖; 納米微球; 微乳液成核; 藥物緩釋; 構象
近年來,在醫學和制藥學領域,人們一直在尋求一種低環境負荷、低能耗、低成本的制造高度功能化納米膠囊的方法.期望納米膠囊可以將各種物質包裹在其內核,且努力使它們的尺寸與典型病毒尺寸相近,使其能夠在血液中長時間循環[1-2],并最終穿透腫瘤附近組織被破壞的毛細血管,達到腫瘤靶向治療的目的[3-4],實現對腫瘤組織的“被動靶向性”.因而納米微粒的制備和性能研究是當前藥物遞送系統的研究熱點[5-7].從體內安全性的角度考慮,人們逐漸寄希望于以天然高分子材料為原料開發聚合物納米膠囊.殼聚糖(CS)由于具有抗潰瘍、防止細菌侵蝕、可生物降解及良好的生物相容性等特性[8-10],近年來得到了越來越多的關注.殼聚糖基納米微粒可使藥物分子順利通過上皮組織,促進藥物的滲透吸收[8,11].Tanima等[12]用交聯法制備了超細殼聚糖納米粒子.經靜脈注射,納米粒子可避開網狀類皮的吞噬,在血液中長時間保留,用放射性元素(99m Tc)示蹤殼聚糖納米粒子在老鼠體內的分布,2 h后仍可在血液中檢測到,同時粒子還分布到心臟、肝臟、腎臟、膀胱、脊柱和骨骼中,說明這種納米粒子可用于骨骼的成像和靶向傳輸.以上研究證實合成穩定、可再生并能負載和包封藥物的殼聚糖納米微粒的可行性及必要性.
具有負載、靶向、控釋等作用的殼聚糖納米微球的制備方法有乳化交聯、溶劑蒸發、形成復乳、噴霧干燥、沉淀析出及復凝聚法等.這些方法需在較苛刻的制備條件如存在有機溶劑、乳化劑和超聲振蕩下進行,因而在實際應用上受到了一定程度的限制[13].
阿司匹林是一種弱酸性藥物,遇濕氣即緩慢水解成為水楊酸,主要用作解熱鎮痛藥和抗風濕藥.研究表明:小劑量的阿司匹林具有較好的抗血栓作用,可用于心血管系統疾病的預防和治療.但普通的阿司匹林片,在體內水解成水楊酸后對胃腸道黏膜有刺激作用.為了減少患者的服藥次數,降低胃腸道的不良反應,使血液中藥物濃度平穩持久,提高藥物療效,可將其制成緩釋微球并通過靜脈注射使用.
本文利用殼聚糖分子在不同pH值的水溶液中構象的變化,采用一種介于沉淀析出法和溶液中離子交聯法的新方式,可稱之為微乳液成核-離子交聯法,使殼聚糖從乳液中以離子交聯納米微粒的形式析出,同時,通過氫鍵相互作用和疏水相互作用,將溶液中的阿司匹林和水楊酸(阿司匹林的水解產物)包埋到微粒之中,制備得到阿司匹林/殼聚糖納米微球,用FTIR、FESEM、TEM、DLLS和XRD對其結構和形貌進行了表征,考察了微粒在注射用生理鹽水和葡萄糖溶液中的釋藥行為,為開發利用可靜脈注射型殼聚糖基納米微粒奠定理論基礎.
1 實驗方法
1.1 原料及試劑
殼聚糖,中國浙江金殼生物化學有限公司,脫乙酰度為90.8%,動力黏度為142 mPa·s,平均聚合度約為350;阿司匹林(實驗室自制),用作藥物模型; Span-80,廣州市潤華食品添加劑有限責任公司精細化工廠,食品級;Tween-80,佛山市科的氣體化工有限公司,食品級;檸檬酸三鈉及其它試劑均為分析純,未經純化直接使用.
1.2 阿司匹林/殼聚糖納米微球的制備
稱取0.20 g殼聚糖于250 mL單口圓底燒瓶中,加入2%(質量分數)醋酸溶液250 mL,開啟電動攪拌裝置緩慢攪拌直至殼聚糖完全溶解.邊攪拌邊加入Span-80 0.4 mL和Tween-80 0.6 mL,繼續攪拌30 min使之形成均勻、透明、穩定的微乳液.接著加入0.26 g經仔細研磨的阿司匹林粉末,高速攪拌使之擴散均勻,然后,緩慢滴加1 mol·L-1的NaOH溶液,同時勻速攪拌,用PHS-3B型精密pH計(中國上海雷磁)在25℃下檢測體系pH值的變化,直至微堿性(pH值為7.20).當體系pH值約為4.30時,阿司匹林完全溶解(水解).NaOH溶液滴加完畢后,中速攪拌并加入0.30 g檸檬酸三鈉,繼續攪拌3 h,使乳液中形成的殼聚糖微球交聯固化.得到的透明乳液用離心機離心分離,上層清液用Lambda 35型紫外-可見分光光度計(美國Perkin Elmer)測定299 nm處阿司匹林的吸光度,下層粘度較大的液體經液氮快速冷凍后用冷凍干燥儀(英國Labconco)冷凍干燥得淡黃色粉末.
1.3 藥物包埋率
1.3.1 阿司匹林標準濃度曲線的制作
準確稱取一定量阿司匹林至100 mL容量瓶中,加去離子水20 mL,溶解后,定容至刻度線,搖勻,配成濃度分別為0.0104、0.0124、0.0144、0.0164、0.0184 mol·L-1的標準溶液.分別取標準溶液用UV-Vis分光光度計測定溶液在299 nm處的吸光度值(A),再以A值對溶液濃度(c)作圖,得線性回歸方程A=70.045c-0.0432,相關系數r2=0.99696.
1.3.2 阿司匹林/殼聚糖納米微粒藥物包封率的測定
將完全干燥的阿司匹林/殼聚糖淡黃色粉末仔細研磨成極細的粉末,精確稱取0.50 g粉末加入50.00 mL的0.1 mol·L-1鹽酸溶液中,攪拌24 h,過濾,精確移取5 mL濾液于50 mL容量瓶中,用0.1 mol·L-1鹽酸稀釋定容至刻度,UV-Vis分光光度計記錄溶液在299 nm處的吸光度,通過標準工作曲線線性回歸方程計算出阿司匹林的含量,按下列公式計算載藥量(L,%)和包封率(R,%)[14].

式中,m1為微球中的阿司匹林質量;m2為稱取的微球質量;m3為投入阿司匹林的總量.
1.4 阿司匹林/殼聚糖納米微粒結構的表征
將冷凍干燥后的淡黃色粉末樣品進行如下測試:紅外光譜在NEXUS 670 FTIR儀(美國Nicolet)上測定,KBr壓片;XRD晶相分析在RU-200B型X射線衍射儀上進行(日本Rigaku公司),Cu Kα輻射,管電流100 mA,管電壓40 kV;淡黃色粉末樣品表面噴金后,用JSM-6701F SEM型場發射掃描電子顯微鏡(FESEM)(日本JEOL)對樣品形貌進行觀察;將樣品超聲分散配制4×10-5g·mL-1的去離子水溶液, 0.1 μm濾膜過濾,用H-600型TEM和BI-200SM型激光光散射儀觀察樣品的平均粒徑及其分布.
1.5 體外釋放實驗
模型藥物的體外釋放實驗按如下方法進行:稱取50 mg阿司匹林/殼聚糖納米微粒于50 mL容量瓶中,加入注射用生理鹽水或其他釋放介質定容后,轉移到100 mL的錐形瓶中,封嚴,在(37.0±0.5)℃的溫度范圍內,分別于1、2、2、3、3、3、5、5、5、5、10、10、10、10、10、20、20、20、20、20、30、30、30、30、30 min量取4 mL緩釋溶液,同時補充4 mL注射用生理鹽水或其他釋放介質,用UV-Vis分光光度計記錄溶液在299 nm處的吸光度值,按線性回歸方程計算釋放t時刻阿司匹林的累積釋放量,并通過下式計算累積釋放分數(Freleased):

式中,mt為釋放t時刻阿司匹林的累積釋放量,m為50 mg樣品中阿司匹林的含量.
2 結果與討論
2.1 溶液pH值變化時殼聚糖分子構象的變化及納米微粒的形成
聚合物的構象是指由于單鍵內旋轉而導致的聚合物分子鏈上的原子和基團在空間的不同排列.殼聚糖是一種多功能的天然陽離子聚電解質,在溶液中的構象一般分為球形、無規線團和剛性棒狀,其構象的變化主要受兩類因素的影響:結構參數,如分子量和脫乙酰度;溶液性質,如溶液離子強度、溶劑、溶液的溫度和pH值等[15].在殼聚糖分子的分子量、脫乙酰度和溶液溫度不變的情況下,改變溶液pH值(同時離子強度也在變化),殼聚糖在溶液中的構象將會發生變化.在酸性溶液中,殼聚糖上的伯氨基(NH2)質子化為NH+3(殼聚糖分子上的大量羥基也會質子化),分子內和分子間存在著較強的靜電排斥作用,此時,殼聚糖分子呈現無規線團狀(如圖1所示).當增加溶液的pH值時(離子強度相應增大),NH+3逐漸解離成NH2,靜電排斥作用減小,此時分子內及分子間氫鍵作用逐漸加強,同時反離子將屏蔽部分未解離的NH+3形成偶極子,分子內和分子間偶極-偶極作用加強,使分子構象收縮蜷曲(圖1中的球形).另外,殼聚糖分子內和分子間的氫鍵作用加強必然導致殼聚糖分子與水分子之間的氫鍵相互作用減弱,而疏水相互作用對殼聚糖分子的構象轉變具有重要的影響.
很多研究者采用熒光光譜[16]、靜態光散射[17-18]、動態光散射[19]等對溶液中殼聚糖的聚集行為進行了研究,結果表明殼聚糖在稀溶液中的臨界聚集濃度(cac)大約是1.0×10-3g·mL-1.本文中殼聚糖的溶液濃度為0.8×10-3g·mL-1,接近于殼聚糖的臨界聚集濃度,因而隨著溶液pH值和離子強度的增大,相應增大的除殼聚糖分子內的氫鍵和偶極-偶極相互作用外,還有分子間的氫鍵及偶極-偶極相互作用,殼聚糖的分子鏈構象由無規線團轉變為球形,同時發生分子間的團聚現象.
隨溶液pH值和離子強度的逐漸增加,殼聚糖分子鏈由較為伸展的無規線團轉變為球形構象,表明分子親水性降低,疏水相互作用加強,而溶液中存在的Tween-80和Span-80對殼聚糖的球形構象有一定的穩定作用.當溶液中加入檸檬酸三鈉時, COO-陰離子與殼聚糖分子鏈上殘留的NH+3之間的靜電相互作用進一步交聯固化了殼聚糖微球(圖2).
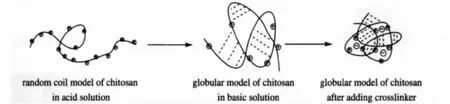
圖1 單分子鏈殼聚糖在不同pH值稀水溶液中的分子構象Fig.1 Molecular conformation of chitosan in dilute solution with different values of pH
2.2 阿司匹林/殼聚糖納米微球的FTIR分析和藥物包封率測定
樣品中各組分間的相互作用能影響分子中化學鍵的鍵長、鍵的方向及強度,從而導致功能基頻率及強度的改變.因此,各官能團對紅外輻射的吸收位置可能會發生較大程度的偏移.
殼聚糖的FTIR光譜如圖3中譜線a所示. 1653.06 cm-1處較強的吸收峰為酰胺I帶(C═O的伸縮振動吸收),1599.85 cm-1處的吸收峰為脫乙酰后NH2的面內彎曲振動吸收,而1310.39及1269.48 cm-1處的弱吸收可以歸屬為酰胺III帶(分別為締合態、游離態未脫乙酰C—N鍵伸縮振動吸收峰).多糖結構的特征吸收帶出現在1156.24 cm-1(吡喃環中C—O—C的不對稱伸縮振動峰),1424.92 cm-1(CH2—OH中O—H的面內彎曲振動特征吸收峰), 1082.51 cm-1(CH2—OH中C—O伸縮振動吸收峰)以及1029.98 cm-1(CH—OH中C—O伸縮振動吸收峰)[20-22].1385.28 cm-1處的吸收可能是由于CH的彎曲振動所致.約3423 cm-1處寬而強的吸收峰可歸屬于糖環上氫鍵締合O—H、N—H的伸縮振動.
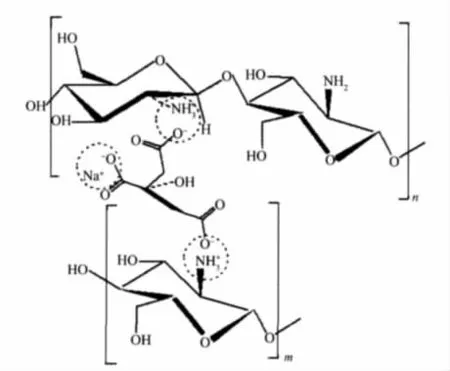
圖2 檸檬酸三鈉交聯殼聚糖的結構示意圖Fig.2 Schematic diagram of the structure of chitosan crosslinked by trisodium citrate
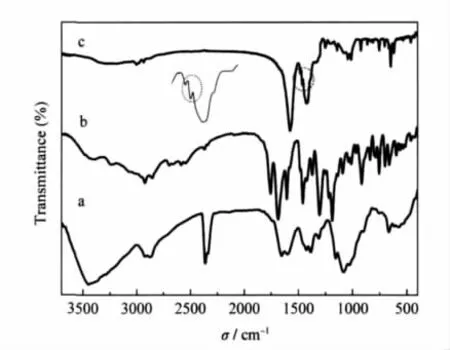
圖3 殼聚糖(a)、阿司匹林(b)和阿司匹林/殼聚糖(c)納米微粒的FTIR光譜圖Fig.3 FTIR spectra of the chitosan(a),aspirin(b), and aspirin/chitosan(c)nanosphere
阿司匹林/殼聚糖納米微球的FTIR光譜如圖3中譜線c所示,由于制備過程為微堿性,殼聚糖水解脫乙酰,1653.06 cm-1處酰胺 I帶、1310.39及1269.48 cm-1處的酰胺III帶相對酰胺II帶減弱[23].而1599.85 cm-1處酰胺II帶向低波數1575.76 cm-1漂移且強度加強,這可能是由于解離NH2與水楊酸中OH之間的氫鍵相互作用以及部分未解離NH+3與檸檬酸三鈉中COO-之間的靜電相互作用使吸收峰位置紅移,同時與水楊酸苯環的C═C伸縮振動疊加使吸收峰強度增大.而3423 cm-1處O—H和N—H寬而強的吸收峰相對減弱并向低波數漂移,再次表明NH2的質子化和氫鍵及靜電相互作用的存在.由于水解及酸堿中和,譜線b中1755.31 cm-1處阿司匹林酯基中的C═O和1685.96 cm-1處芳香羧酸中C═O的伸縮振動特征吸收在阿司匹林/殼聚糖譜圖中消失.而可以用于鑒定苯環是否存在的C═C伸縮振動的四個譜帶是1625-1580 cm-1、1590-1570 cm-1、1525-1470 cm-1和1460-1440 cm-1,其中在阿司匹林/殼聚糖譜圖中可以明顯觀察到位于1481.92、1463.67 cm-1處的兩個吸收峰,與殼聚糖在1423.05 cm-1的CH2—OH中OH的面內彎曲振動特征吸收峰及1386.78 cm-1處的CH的彎曲振動吸收峰部分重疊,表明殼聚糖納米微球中水楊酸成分的存在.另一個應用較多可以鑒定苯環取代類型的吸收峰為754.96 cm-1處的峰,為苯環上四個相鄰C—H鍵的伸縮振動吸收峰.水楊酸中COO-基團的不對稱與對稱伸縮振動吸收峰分別出現在1610-1550 cm-1及1440-1360 cm-1范圍內,這也是譜線c中1575.76與1423.27 cm-1處吸收峰較殼聚糖的FTIR譜線a強度增大的另一個原因,進一步說明在微乳液成核-離子交聯過程中,殼聚糖從乳液中以離子交聯納米微粒形式析出的同時,通過氫鍵相互作用及疏水相互作用,溶液中的水楊酸(阿司匹林的水解產物)也被成功包埋到微粒之中,制備得到了阿司匹林/殼聚糖納米微球.
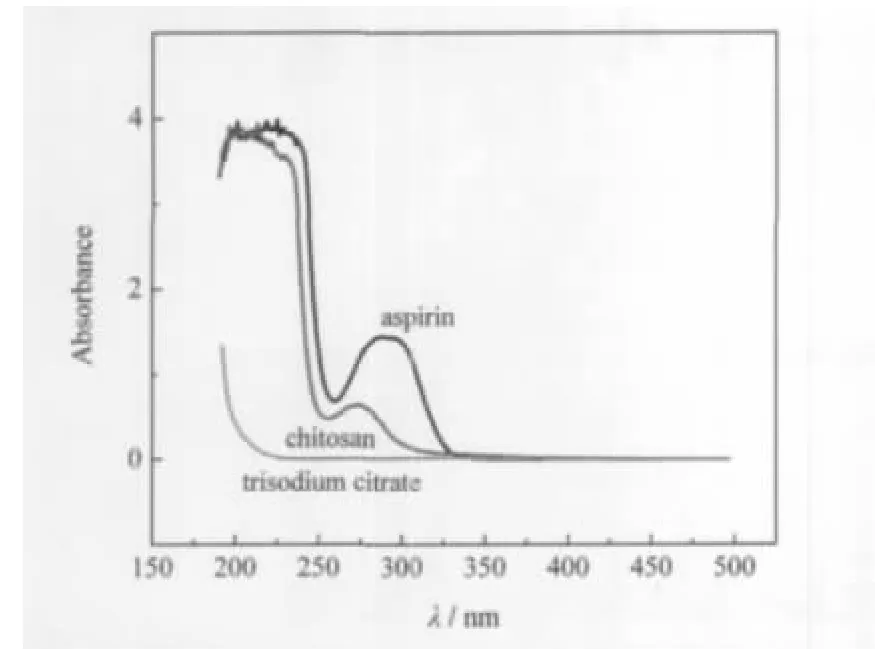
圖4 水溶液中阿司匹林、殼聚糖和檸檬酸三鈉的UV-Vis光譜圖Fig.4 UV-Vis spectra of aspirin,chitosan,and trisodium citrate in aqueous solution
制備過程投料比中阿司匹林的用量約為殼聚糖總量的130%,高速離心后上層清液用UV-Vis分光光度計檢測其中所含阿司匹林在299 nm處的吸光度值(圖4),代入標準濃度曲線的線性回歸方程,求得高速離心后上層清液中阿司匹林的含量約為0.15 g,約占阿司匹林投入量的58%.下層粘度較大的液體經冷凍干燥后得到阿司匹林/殼聚糖淡黃色粉末,研磨后稱取0.50 g粉末加入50 mL 0.1 mol·L-1的HCl溶液,攪拌24 h,過濾,稀釋,UV-Vis分光光度計記錄溶液在299 nm處的吸光度值,通過標準濃度曲線線性回歸方程,按公式(1)、(2)計算得載藥量約為55%,包封率約為42%.
由于阿司匹林在微堿性溶液中易溶解,導致藥物包封率較低.針對微堿性環境中溶解度較小的藥物(如抗腫瘤藥物紫杉醇)或在微堿性溶液中穩定的氨基酸、肽和蛋白質等新型藥物,藥物的包封率應會相應提高,而這種微乳液成核-離子交聯法制備的殼聚糖納米微粒為氨基酸、肽和蛋白質的輸送提供了新的途徑,在方便病人用藥和被吸收進入細胞達到細胞內靶的方面具有潛在的價值.
2.3 阿司匹林/殼聚糖納米微粒的結晶形態及形貌分析
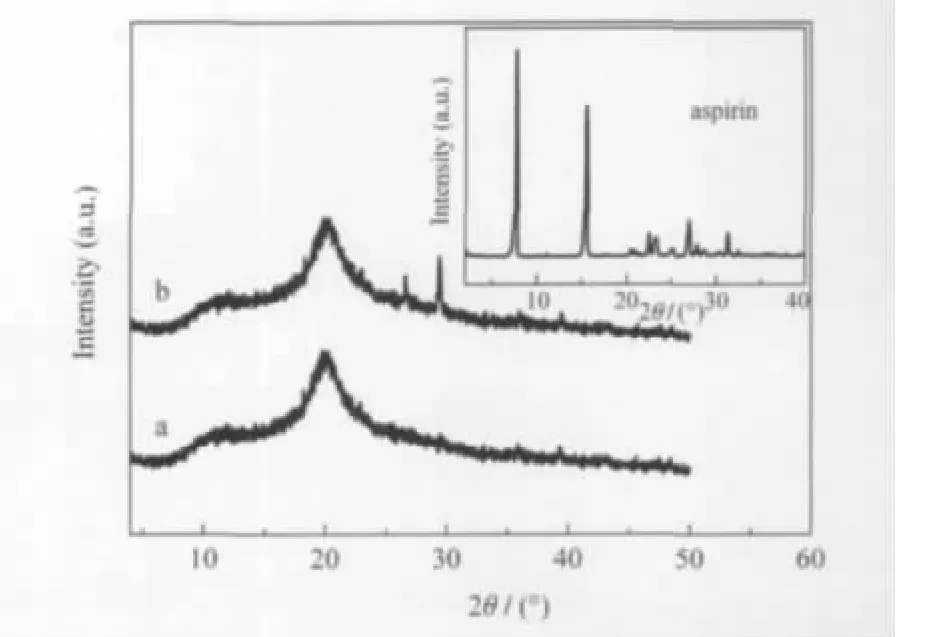
圖5 殼聚糖(a),阿司匹林/殼聚糖(b)和阿司匹林(插圖)的XRD譜圖Fig.5 XRD patterns of chitosan(a),aspirin/chitosan (b),and aspirin(inset)
圖5 為殼聚糖和阿司匹林/殼聚糖的XRD譜圖.由圖5譜線a可以看出,殼聚糖具有一定的結晶性,在2θ為11.9°(100)和20.2°(020)附近有兩個較強的基面衍射峰,但衍射峰強度較弱,寬度較大,說明其結晶度較低,與文獻報道[24-25]類似.阿司匹林/殼聚糖(圖5譜線b)與殼聚糖的XRD譜圖基本一致,說明微乳液成核-離子交聯過程對殼聚糖的晶體結構和結晶度沒有影響,同時也說明阿司匹林對殼聚糖的結晶幾乎沒有影響.然而,在2θ為26.46°,29.22°處觀察到兩個弱衍射峰,目前無法將其進行正確解釋,推測可能是因為在微乳液成核-離子交聯過程中,隨著溶液pH值和離子強度的增大,殼聚糖分子內和分子間的氫鍵和偶極-偶極相互作用也隨著增大,而大分子這種長程有序的協同相互作用促使殼聚糖分子鏈局部排列規整,導致其晶體結構發生微小的變化.圖5插圖中的譜線顯示阿司匹林在衍射角2θ為7.74°和15.52°等處有強的衍射峰[26-27].而這些特征衍射峰在譜線b中沒有出現,表明由于氫鍵和疏水相互作用阿司匹林在殼聚糖載體上高度分散,分子間未形成堆砌,阿司匹林主要以無定形態存在于納米微粒中.
采用場發射掃描電子顯微鏡觀察未交聯殼聚糖和阿司匹林/殼聚糖的形貌特征,結果分別示于圖6a和6b.從圖6a中可以看出,在堿性環境中,分子內及分子間氫鍵和偶極-偶極作用加強,促使殼聚糖分子構象收縮蜷曲成球形.但由于未加離子交聯劑,粒子表面粗糙.而分子間的氫鍵及偶極-偶極相互作用使團聚現象發生.而圖6b顯示的結果表明,利用微乳液成核-離子交聯法制得的阿司匹林/殼聚糖呈比較規整的球型,微球的平均直徑小于100 nm,較未交聯殼聚糖粒子的直徑大,而且表面光滑,粒徑分布均勻,粘連團聚現象較弱.阿司匹林的加入是粒徑增大的主要原因,而阿司匹林和檸檬酸三鈉對殼聚糖分子的交聯作用可能使粒子表面更為光滑且團聚減輕.

圖6 未交聯殼聚糖(a)和阿司匹林/殼聚糖(b)的場發射掃描電鏡照片Fig.6 FESEM images of uncrosslinked chitosan(a)and aspirin/chitosan(b)
制樣過程中的離心分離使溶液濃度局部增大也可能導致團聚,所以在低濃度下可能得到分散狀態較好的阿司匹林/殼聚糖微球.將阿司匹林/殼聚糖超聲分散到去離子水中,樣品在水中的質量濃度為4×10-5g·mL-1,分別用透射電子顯微鏡和動態激光光散射觀察阿司匹林/殼聚糖在去離子水中的分散情況,并測定粒徑及粒徑分布,結果分別示于圖7和圖8.從圖中可以看出,阿司匹林/殼聚糖顆粒均勻且分散性良好,平均粒徑約88 nm.
2.4 阿司匹林/殼聚糖納米微粒的體外釋藥性能
殼聚糖分子的構象以及檸檬酸三鈉、殼聚糖和阿司匹林的電荷密度與溶液pH值密切相關,因而溶液pH值變化會影響到阿司匹林/殼聚糖納米微粒的交聯密度的變化,從而影響藥物釋放速率.本文考察了阿司匹林/殼聚糖納米微粒在pH值分別為1.8和7.2的緩沖溶液中的釋放行為,結果表明,在pH值為7.2的緩沖溶液中,阿司匹林/殼聚糖保持收縮狀態,藥物釋放很慢,250 min內釋藥僅23%.而在pH值為1.8的緩沖溶液中,由于檸檬酸三鈉的交聯作用消失,殼聚糖分子構象伸展,250 min內藥物釋放已近90%.與Shu等[28]得到的結果一致.
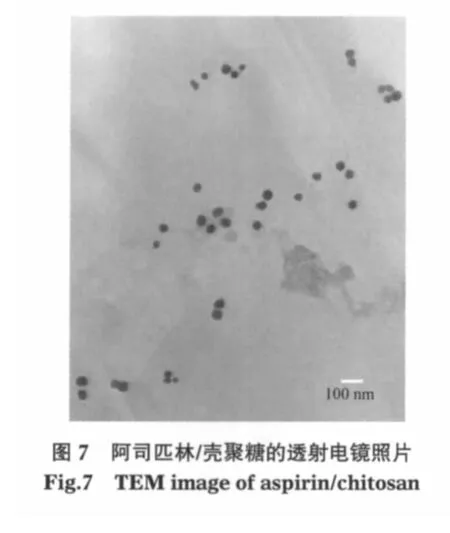
圖9所示的是去離子水、注射用生理鹽水及葡萄糖溶液中阿司匹林/殼聚糖納米微粒累積釋放量隨時間的變化曲線.不難看出,在釋放的初始階段,葡萄糖溶液中阿司匹林/殼聚糖納米微粒的藥物釋放速率明顯小于生理鹽水中釋放的速率,40 min內藥物釋放不到50%,而在生理鹽水中釋放已達到80%.吸附在微粒表面的藥物擴散可能是導致釋放初期快速釋藥的重要因素.經過一個突釋階段后,釋放速度減慢,釋藥約400 min,生理鹽水中阿司匹林/殼聚糖納米微粒釋放率約為95%,葡萄糖溶液中釋放率約為55%,仍有45%的藥物未釋放,納米微球表現出良好的緩釋性能.與生理鹽水中阿司匹林的釋放速率相比,去離子水中阿司匹林/殼聚糖納米微粒的釋放比較緩慢,與葡萄糖溶液中的釋放接近.
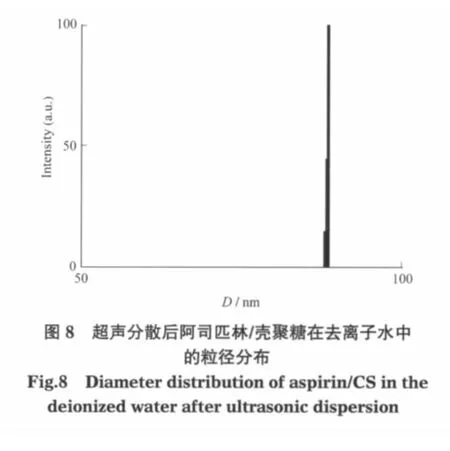
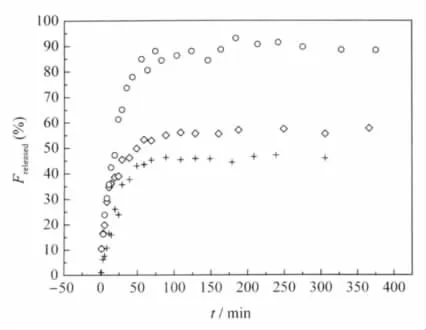
圖9 生理鹽水(○)、葡萄糖溶液(◇)和去離子水(+)中阿司匹林/殼聚糖納米微粒的累積釋放量隨時間的變化曲線(37℃)Fig.9 Release profiles of aspirin/CS nanospheres in saline(○),glucose solution using in medicine(◇)and deionized water(+)at 37℃
研究大分子聚電解質的溶液行為,離子強度是一個關鍵的影響因素[29].人們已深入地研究了離子強度對殼聚糖溶液性質和構象的影響.Tsaih等[30]對稀溶液中離子強度對殼聚糖分子構象影響的研究發現,隨著溶液離子強度的增加,殼聚糖的分子構象從伸展變得蜷曲.在低離子強度時,第三電粘度(the third electroviscous)的影響起主要作用,殼聚糖呈現伸展的分子構象.在高離子強度時,反離子對質子化氨基的屏蔽作用使分子蜷曲.當離子強度增加到無窮大時,所有的質子化氨基都被反離子中和,分子內的靜電斥力消失,所有的殼聚糖分子無論分子量大小都變成緊縮的球形,當用脲將分子內的氫鍵消除時,殼聚糖分子又變得伸展.然而,對于殼聚糖基于靜電相互作用和氫鍵相互作用形成的納米微球,當溶液中加入小分子電解質(例如NaCl)時,電解質對殼聚糖構象的影響研究較少.本文中圖9所示的實驗結果表明,NaCl溶液中阿司匹林的釋放速率更快,表明在生理鹽水中殼聚糖的離子交聯密度可能較低,殼聚糖的構象可能由蜷曲的球變化為伸展的蠕蟲狀鏈,從而使藥物釋放.這可能是因為小分子電解質破壞了殼聚糖納米粒的水合層,由于滲透壓的關系由水溶液向粒子內部滲透,與殼聚糖的質子化氨基相互競爭,Na+與檸檬酸鹽COO-之間的靜電相互作用部分取代了殼聚糖鏈上NH+3與檸檬酸鹽COO-之間的靜電相互作用,導致低的交聯密度[31].同時,更為重要的是,NaCl的滲入,削弱了殼聚糖分子間和分子內的氫鍵作用,有可能導致殼聚糖的構象由蜷曲變化為伸展的蠕蟲狀鏈,因而使阿司匹林的釋放速率由于小分子電解質的加入而加快.可見,小分子電解質對殼聚糖溶液行為的影響,到目前為止,仍有高分子相轉變現象、相轉變機理、相平衡原理等諸多問題,等待廣大高分子工作者進行深入細致的研究.
3 結 論
場發射掃描電子顯微鏡、透射電子顯微鏡和動態激光光散射研究結果表明微乳液成核-離子交聯法不失為一種制備殼聚糖基納米載藥微球的簡單易行而又有效的方法.UV-Vis分光光度計對納米微球在注射用生理鹽水和葡萄糖溶液中的釋藥行為跟蹤結果表明,實驗條件下葡萄糖溶液中殼聚糖基納米載藥微球具有良好的藥物緩釋作用,而生理鹽水中較快的釋放速率可能歸因于小分子鹽對殼聚糖分子鏈中氫鍵和靜電相互作用的破壞.小分子電解質對殼聚糖在溶液中的構象轉變的這種影響有待于進一步研究.
1 Yang,Z.;Zheng,S.Y.;Harrison,W.J.;Harder,J.;Wen,X.X.; Gelovani,J.G.;Qiao,A.;Li,C.Biomacromolecules,2007,8:3422
2 Aliabadi,H.M.;Brocks,D.R.;Lavasanifar,A.Biomaterials, 2005,26:7251
3 Greish,K.;Sawa,T.;Fang,J.;Akaike,T.;Maeda,H.J.Control. Release,2004,97:219
4 Lee,E.S.;Na,K.;Bae,Y.H.J.Control.Release,2005,103:405
5 Attwood,D.;Booth,C.;Yeates,S.G.;Chaibundit,C.;Ricardo,N. M.P.S.Int.J.Pharm.,2007,345:35
6 Yang,Z.L.;Yang,K.W.;Li,X.R.;Liu,Y.Chin.Pharm.J.,2007, 42:519 [楊卓理,楊可偉,李馨儒,劉 艷.中國藥學雜志, 2007,42:519]
7 Rijcken,C.J.;Snel,C.J.;Schiffelers,R.M.;van Nostrum,C.F.; Hennink,W.E.Biomaterials,2007,28:5581
8 Calvo,P.;Remu?án-López,C.;Vila-Jato,J.L.;Alonso,M.J. J.Appl.Polym.Sci.,1997,63:125
9 Bravo-Osuna,I.;Ponchel,G.;Vauthier,C.Eur.J.Pharm.Sci., 2007,30:143
10 Peppas,N.A.;Hilt,J.Z.;Khademhosseini,A.;Langer,R.Adv. Mater.,2006,18:1345
11 Xu,Y.;Du,Y.Int.J.Pharm.,2001,250:215
12 Tanima,B.;Susmita,M.;Singh,K.Int.J.Pharm.,2002,243:93
13 Huang,X.L.;Zhang,L.M.J.Function.Polym.,2003,16:594 [黃小龍,張黎明.功能高分子學報,2003,16:594]
14 He,Q.F.;Li,G.M.;Wu,H.Z.;Lu,Z.M.Chin.J.Appl.Chem., 2004,21:192 [何強芳,李國明,巫海珍,盧志敏.應用化學, 2004,21:192]
15 Sorlier,P.;Viton,C.;Domard,A.Biomacromolecules,2002,3: 1336
16 Amiji,M.M.Carbohyd.Polym.,1995,26:211
17 Anthonsen,M.W.;V?rum,K.M.;Hermansson,A.M.;Smidsrod, O.;Brant,D.A.Carbohyd.Polym.,1994,25:13
18 Schatz,C.;Pichot,C.;Delair,T.;Viton,C.;Domard,A.Langmuir, 2003,19:9896
19 Buhler,E.;Rinaudo,M.Macromolecules,2000,33:2098
20 Mladenovska,K.;Cruaud,O.;Richomme,P.;Belamie,E.;Raicki, R.S.;Venier-Julienne,M.C.;Popovski,E.;Benoit,J.P.; Goracinova,K.Int.J.Pharm.,2007,345:59
21 Mi,F.L.;Sug,H.W.;Shyu,S.S.Carbohydr.Polym.,2002,48: 61
22 Li,J.F.;Zhang,L.;Li,J.F.;Zou,Q.;Yang,W.H.;Li,Y.B.Chem. J.Chin.Univ.,2008,29:1874 [李峻峰,張 利,李鈞甫,鄒 琴,楊維虎,李玉寶.高等學校化學學報,2008,29:1874
23 Ren,D.W.;Wang,Y.L.;Bao,D.C.;Xie,W.Y.;Ma,X.J. Spectros.Spectr.Analy.,2006,26:1825 [任東文,王一力,包德才,謝威揚,馬小軍.光譜學與光譜分析,2006,26:1825
24 Kim,J.H.;Lee,Y.M.Polymer,1993,34:1952
25 Zeng,R.;Tu,M.;Liu,H.W.;Zhao,J.H.;Zha,Z.G.;Zhou,C.R. Carbohydr.Polym.,2009,78:107
26 Yamada,H.;Teradac,K.;Suryanarayanana,R.J.Pharm. Biomedic.Anal.,2010,51:952
27 Phadnis,N.V.;Cavatur,R.K.;Suryanarayanan,R.J.Pharm. Biomedic.Anal.,1997,15:929
28 Shu,X.Z.;Zhu,K.J.Int.J.Pharm.,2002,233:217
29 Gao,Q.;Wang,G.J.;Li,W.T.Chemistry,2009,72:1 [高 群,王國建,李文濤.化學通報,2009,72:1]
30 Tsaih,M.L.;Chen,R.H.Int.J.Biol.Marcromol.,1997,20:233
31 Shu,X.Z.;Zhu,K.J.;Song,W.H.Int.J.Pharm.,2001,212:19
Preparation and Release Behavior in vitro of Aspirin/Chitosan Nanospheres by Nucleation and Ionic Crosslinking in Emulsion
JIN Shu-Ping FENG Lei*HE Wen-Yan HAN Yu-Qi WEI Yu-Juan
(Key Laboratory of Resources and Environmental Chemistry of West China,Department of Chemistry,Hexi University, Zhangye 734000,Gansu Province,P.R.China)
Chitosan(CS)nanosphere loaded aspirin(aspirin/CS)was prepared by nucleation and ionic crosslinking in an emulsion used for medical and pharmaceutical applications.Chemical component,morphology,size distribution, andcrystalstructureofnanosphereswerecharacterizedbyFouriertransforminfrared(FTIR)spectroscopy,fieldemission scanning electron microscopy(FESEM),transmission electron microscopy(TEM),dynamic laser light scattering (DLLS),and X-ray powder diffraction(XRD).Results showed that the diameter of a typical aspirin/CS nanosphere is about 88 nm and the distribution is uniform.The crystal structure of CS does not change during the nucleation process. The crystallinity of aspirin is dramatically reduced and aspirin is almost amorphous in the nanosphere.The drug content(mass fraction),the drug loading efficiency,and the in vitro release profiles under different conditions were investigated using UV-Vis spectrophotometry.Results showed that the drug content was about 55%,the drug loading efficiency reached 42%,and the chitosan nanosphere displayed an excellent drug-controlled release behavior under the experimental conditions.
Chitosan;Nanosphere;Nucleation in emulsion;Drug-controlled release;Conformation
O645
Received:January 5,2010;Revised:April 26,2010;Published on Web:July 5,2010.
*Corresponding author.Email:flgkl@163.com,zjxjsp@163.com;Tel:+86-936-8282066
?Editorial office of Acta Physico-Chimica Sinica
猜你喜歡
河北科技師范學院學報(2022年2期)2022-08-26 08:55:40
河北科技師范學院學報(2021年1期)2021-05-10 03:34:20
中成藥(2017年12期)2018-01-19 02:06:57
電源技術(2017年1期)2017-03-20 13:37:59
廣西科技大學學報(2016年1期)2016-06-22 13:10:38
天然產物研究與開發(2016年1期)2016-06-05 10:29:25
食品界(2016年4期)2016-02-27 07:36:46
中國果菜(2015年2期)2015-03-11 20:01:01
應用化工(2014年7期)2014-08-09 09:20:21
應用技術學報(2014年4期)2014-02-28 14:52:40
