
鳥嘌呤四鏈體中Na+的移動
2010-11-30 10:56:10楊忠志
物理化學學報 2010年2期
郭 慈 劉 翠 楊忠志
(遼寧師范大學化學化工學院,遼寧大連 116029)
EvdW和Eelec是非鍵作用勢能.EvdW描述非鍵原子間的vdW相互作用,表達式為:
鳥嘌呤四鏈體中Na+的移動
郭 慈 劉 翠 楊忠志*
(遼寧師范大學化學化工學院,遼寧大連 116029)
Na+-G-四鏈體復合物是一個明顯的極化體系,其形成或解離過程中,Na+的移動路線目前還不十分明確. σπ水平的原子-鍵電負性均衡方法融合進分子力學(ABEEMσπ/MM)模型除原子位點外,還明確地定義了孤對電子、σ鍵和π鍵的位置,并且各位點電荷隨分子環境改變而浮動,因此能更好地反映該體系的極化現象.本文應用ABEEMσπ/MM方法研究了Na+-G-四平面復合物的性質,包括它的幾何構型、電荷分布和結合能等,并在MP2/6-31G(d,p)水平上做了相應的從頭算,兩種結果十分吻合.Na+的存在改變了G-tetrad的氫鍵方式.通過比較Na+各條移動路線中體系的結合能,預測G-四鏈體中三個Na+最有可能沿α方向依次移出.以上研究為進一步應用ABEEMσπ/MM模型進行G-四鏈體中離子交換通道的動力學模擬打下堅實的基礎.
G-四鏈體;Na+-G-四平面;ABEEMσπ/MM方法;從頭算;移動路線
端粒是染色體末端一種特殊的、富含鳥嘌呤的結構.多項研究[1-7]發現,在某些離子條件下,富含鳥嘌呤端粒的尾鏈,可以通過分子內折疊或分子間相互作用,形成特殊的DNA二級結構,即鳥嘌呤四鏈體(G-quadruplex,G4).此結構由三個平行排列的G-四平面堆疊而成;每個G-四平面(G-tetrad)由4個鳥嘌呤通過分子間氫鍵連接構成,離子存在于G4空穴中.G4的形成與端粒酶引起的端粒延長有關,可抑制端粒酶發揮作用,干擾端粒復制,破壞端粒功能,對細胞生長、分化、凋亡等基本生命活動產生影響[8-9],G4的特殊結構與性質使之成為藥物設計的理想方向[10-15].
已有文獻[16-19]對G4進行了研究,包括結構、穩定性和G4中離子交換等相關問題.研究表明:與金屬離子相互作用對G4的形成至關重要,離子的作用就是穩定G4.G4的穩定性和結構隨它含有的離子類型而定,也就是說,G4對離子具有選擇性.Gu等[20]研究了G4對離子的選擇,表明在氣相中,單價陽離子穩定G4的順序是Li+>Na+>K+,而在水溶液中順序恰好相反.Ma等[21]研究了親脂性的G4中離子交換的問題,指出:并不是所有離子的結合位置都是均等的.然而,G4形成或解離過程中離子的移動路線目前還不是十分明確.要判斷G4中離子的移動路線,必須要研究離子與G4的相互作用.對此本文應用從頭算(MP2)[22]和ABEEMσπ/MM[23-30]方法,以一個Na+和1片G-tetrad(Na+-G-tetrad)的相互作用為切入點,驗證了ABEEMσπ/MM方法的準確性.在此基礎上,應用ABEEMσπ/MM方法研究了兩片G-tetrad和Na+(Na+-G-tetrads)的相互作用,通過比較Na+移動過程中體系的結合能,預測了G4中Na+的移動路線.
1 計算方法
1.1 從頭計算方法
在量子化學計算方法中,通常用來計算能量的有HF、DFT、MP2及CCSD[31]等方法.其中HF方法沒有考慮電子相關效應,計算出的能量不夠準確; DFT方法雖考慮了電子相關效應,但不能很好地處理弱鍵相互作用;CCSD方法雖然計算能量比較準確,但由于計算機條件限制,只能做十幾個原子的體系.G4是一個相對較大的體系,考慮到MP2方法的計算精度,以及該方法節省計算機時,本文采用MP2方法計算能量,基組為6-31G(d,p).由于本文的初始構型是實驗構型,因此沒有進行幾何優化,在計算能量的過程中應用 counterpoise方法進行了BSSE校正.
1.2 ABEEMσπ/MM模型
ABEEMσπ力場中,分子的總能量EABEEMσπ表示為:
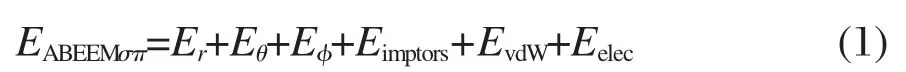
其中Er是鍵伸縮振動勢能;Eθ是分子中連續鍵連的三個原子形成的鍵角彎曲振動勢能;Eφ和Eimptors是分子中連續鍵連的四個原子形成的二面角扭轉勢能和非共面扭轉勢能.它們的表達式如式(2-5)所示:

其中kr和kθ表示鍵伸縮和鍵角彎曲勢能的力常數, r、θ和φ是實際的鍵長、鍵角和二面角值,req和θeq表示平衡鍵長和鍵角值,V1、V2、V3和ν為二面角扭轉勢能項及非共面扭轉勢能項的展開力常數.
EvdW和Eelec是非鍵作用勢能.EvdW描述非鍵原子間的vdW相互作用,表達式為:

f為常數,ε為勢阱深,σ為碰撞直徑.此處采用了標準的聯合規則:εij=(εiiεjj)1/2,σij=(σiiσjj)1/2.對于分子內相互作用,任何1-2和1-3關系的i-j原子對,fij=0.0,任何1-4關系的i-j原子對,fij=0.5,所有其它情況下fij=1.0.
靜電相互作用項 Eelec中的電荷計算是ABEEMσπ模型的精華所在,公式如下:

其中,qi和qj是位點i和j的電荷,rij是位點i和j的距離,當i和j之間的最短連接路徑關系(包括σ鍵位點)小于1-6時,kij=0;當i和j在氫鍵相互作用區域時,kij=kH-bond(氫鍵擬合函數);其它所有情況,kij= 0.57.在ABEEMσπ模型中,σ鍵處于兩成鍵原子共價半徑之比處;π鍵處于垂直于雙鍵所在平面,置于雙鍵原子上下兩側共價半徑處;孤對電子處于距離雙鍵原子共價半徑處.
1.3 ABEEMσπ/MM模型參數的確定
ABEEMσπ參數的擬合是計算所有性質的一個關鍵步驟.本文通過線性回歸和最小二乘法優化確定參數.參數的擬合不僅產生與從頭計算相一致的電荷分布、結合能,而且獲得了與實驗構型相一致的幾何構型[32].其中G4體系的范德華參數列于表1.

表1 ABEEMσπ/MM模型中G4體系的vdW參數Table 1 vdW parameters of G4 in ABEEMσπ/ MM model
如果想要解釋廣泛的化學現象,那么基于靜電力場的分子模擬中,包含氫鍵效應是至關重要的. G4體系中鳥嘌呤間有兩種類型的氫鍵:(1)O6(原子編號見圖1中所示)原子上的孤對電子與其相鄰鳥嘌呤上H45原子之間形成的氫鍵,或O6原子上的孤對電子與其相鄰鳥嘌呤上H47原子之間形成的氫鍵;(2)N3原子上的孤對電子與其相鄰鳥嘌呤上H47原子之間形成的氫鍵.這兩種類型的氫鍵擬合函數是沿用ABEEMσπ/MM模型原有的函數[29].本文特別關注的是Na+與鳥嘌呤中O6原子、N10原子、N3原子上孤對電子之間的靜電相互作用.為了更好地描述這三種靜電相互作用,我們用ABEEMσπ/MM模型擬合了在MP2/6-31G(d,p)水平上的兩個靜電區域處于不同距離時的結合能,使二者結果有很好的一致性.從而引入三個可調參數k(RlpO=,Na+),k(RlpN10,Na+)和k(RlpN3,Na+).這些參數隨Na+與孤對電子之間距離的變化而改變,更好地體現了該區域內的極化現象,這三個參數的表達式分別為:

其中R表示Na+與孤對電子之間的距離.
Na+與G-tetrad相互作用研究中用到的其它參數沿用Yang等人已經報道的相應參數[29-30].
2 結果與討論
2.1 幾何構型和電荷分布
應用ABEEMσπ/MM模型優化的G-tetrad與Na+-G-tetrad結構相差很大.G-tetrad構型中,四個鳥嘌呤通過交叉型氫鍵存在,如圖1(a)所示.Na+-G-tetrad構型中,四個鳥嘌呤趨近于在一個平面內,采用平行型的氫鍵,Na+在四個鳥嘌呤形成空穴的下方,如圖1(b)所示.分析可得,G-tetrad結構中,中心部位的四個氧原子之間存在很大的靜電排斥作用,為了降低這種排斥作用,O6原子上的孤對電子與N40上的H45以及N42上的H47形成了交叉型的氫鍵.當有Na+存在的情況下,Na+中和了G-tetrad中心部位集中的大量負電荷,平衡了G-tetrad中心部位四個氧原子之間的靜電作用,使G-tetrad采取平行型的氫鍵,增加了G-tetrad結構的穩定.
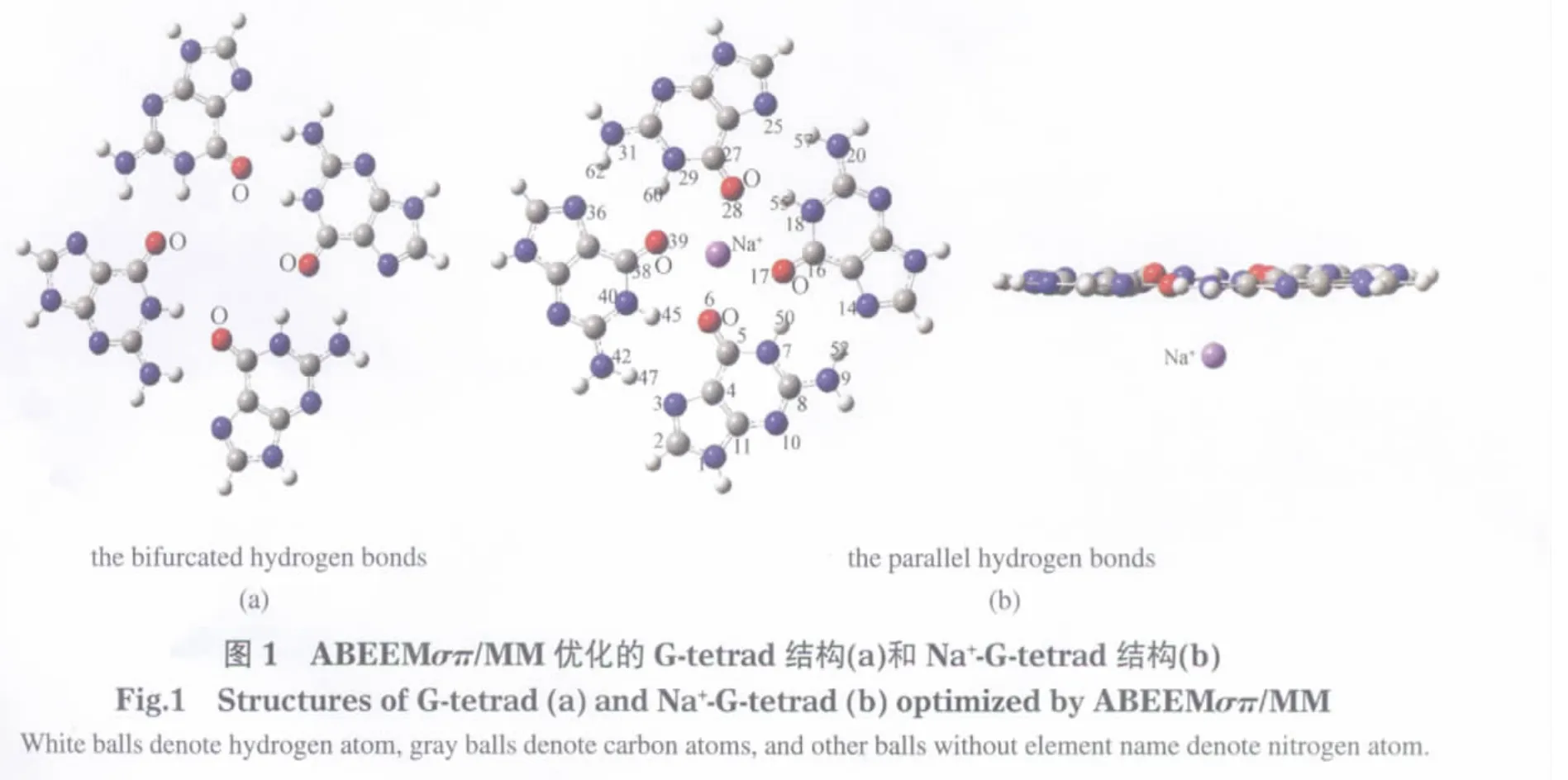

表2 ABEEMσπ/MM模型優化的Na+-G-tetrad的結構參數與實驗數據的比較Table 2 Comparison of structural parameters for Na+-G-tetrad from experiment and optimized by ABEEMσπ/MM model
表2列出的是ABEEMσπ/MM模型優化的Na+-G-tetrad的部分結構參數與實驗構型的比較. ABEEMσπ/MM模型優化得到的Na+-G-tetrad復合物的幾何構型參數與實驗結構參數[32]比較可知,二者的結構符合得很好.鳥嘌呤單體鍵長的絕對平均偏差為0.0013 nm,鍵角的絕對平均偏差為1.18°.從表2中可以看出,原子間距離的絕對平均偏差為0.0026 nm,角度的絕對平均偏差為2.18°,都在較合理的范圍內.可見,ABEEMσπ/MM模型能夠正確地預測Na+-G-tetrad的結構信息.
表3列出的是ABEEMσπ/MM模型計算的G-tetrad和Na+-G-tetrad中氧原子各位點所帶的電荷. Na+-G-tetrad是一個明顯的極化體系,ABEEMσπ/ MM模型除原子位點外,還明確地定義了孤對電子、σ鍵和π鍵的位置,并且隨分子環境的改變,各位點的電荷發生相應的浮動,這樣更好地反映了該體系的極化現象.將表中各位點的電荷回歸到原子后:G-tetrad中,四個氧原子所帶的電荷相等都是-0.5798e;Na+-G-tetrad中,qO6=-0.6538e;qO17= -0.6597e;qO28=-0.6623e;qO39=-0.6450e.由此可以看出,將G-tetrad和Na+-G-tetrad中各位點電荷回歸到原子后,氧原子帶有不同程度的負電荷.因為Na+-G-tetrad結構中Na+帶有一定的正電荷,并且Na+和氧原子間存在靜電相互作用,所以氧原子被極化,帶更多的負電荷.通過對比可知,O的孤對電子上的電荷改變最大,而不是原子位點處.并且有Na+存在時,靠近Na+的π鍵位點的負電荷明顯地比遠離Na+的π鍵位點的負電荷多;沒有Na+存在時O原子上下的π鍵位點所帶電荷基本相等.
ABEEMσπ/MM模型不僅定義了非原子位點,并且引入可調參數來研究Na+與O6原子、N10原子、N3原子上的孤對電子之間的靜電相互作用,所以該模型能很好地模擬G-tetrad和Na+-G-tetrad體系中的氫鍵和靜電相互作用.總體來說, ABEEMσπ/MM模型能夠合理地預測G-tetrad的結構,與其它文獻報道一致[20],并且優化的Na+-G-tetrad構型與實驗構型有很好的一致性,進而說明了ABEEMσπ/MM模型在描述G-tetrad和Na+-G-tetrad的幾何構型方面十分可靠.
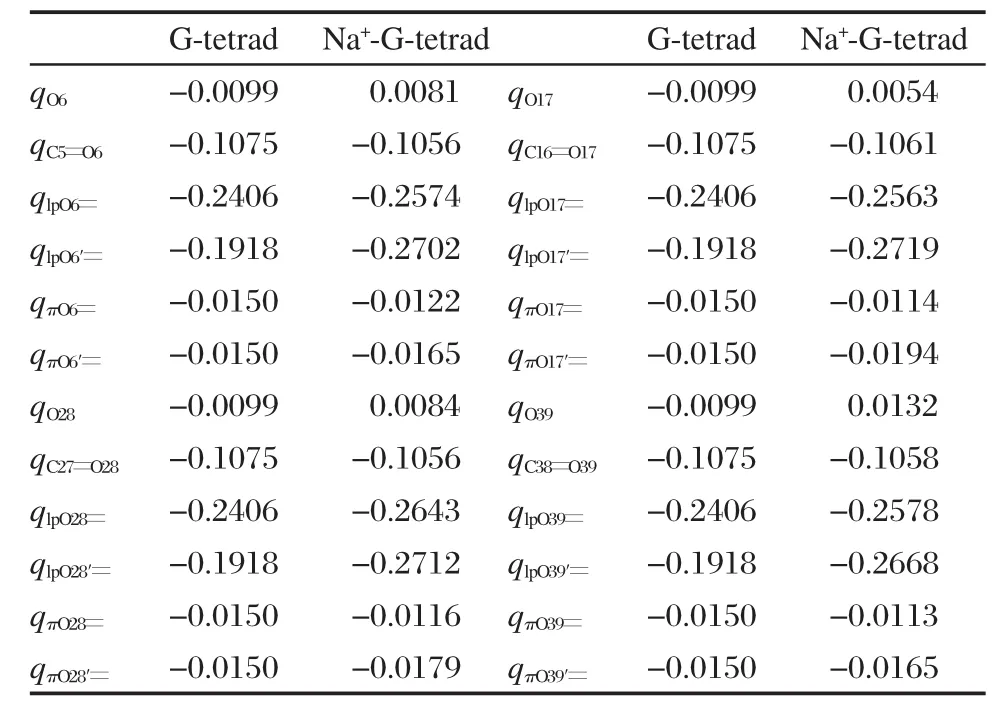
表3 ABEEMσπ/MM計算的G-tetrad和Na+-G-tetrad中氧原子各位點所帶的電荷Table 3 Charges of all sites of O atoms in G-tetrad and Na+-G-tetrad computed by ABEEMσπ/MM

圖2 Na+移動方向正視圖(a)和俯視圖(b)Fig.2 The front(a)and vertical(b)view of the moving direction of Na+
2.2 Na+和一片G-tetrad相互作用
G4形成或解離過程中,涉及到Na+能否移出G4,以及Na+在移動的過程中能否跟其它的離子交換(如K+)等問題,本文應用ABEEMσπ/MM模型初步預測了Na+的移動路線.
初始構型取自蛋白質晶體數據庫,其數據庫標號為1KF1[32].刪去磷酸和糖環,并在堿基上加上氫原子,將其中的鉀離子改為鈉離子.為了檢驗模型的準確性,取第1片G-tetrad和Na+(2)(Na+(2)-G-tetrad)作為初始結構,如圖2所示.討論了一個Na+與一片G-tetrad的相互作用,并與MP2/6-31G(d,p)水平的從頭算結果進行了對比.將Na+(2)和Na+(3)的連線方向定義為α方向;過Na+(2),且垂直于α方向取一個平面I,將第1片G-tetrad中兩鳥嘌呤的夾縫方向在平面I中的投影定義為β方向;將第1片G-tetrad中兩鳥嘌呤的對角線方向在平面I中的投影定義為γ方向,圖中箭頭所指方向為正方向.根據Na+(2)-G-tetrad結構的特點,α、β、γ三個方向是該結構中最具代表性的方向,以Na+(2)所在位置為起點(平衡位置),討論了該離子分別沿α、β、γ三個方向的移動.結合能ΔE計算公式如下:
ΔE=E[Na+(2)]+E[G-tetrad]-E[Na+(2)-G-tetrad](11)其中E[Na+(2)]為Na+(2)的能量,E[G-tetrad]為一片G-tetrad復合物的能量,E[Na+(2)-G-tetrad]為Na+(2)-G-tetrad復合物的能量.
2.2.1 Na+(2)-G-tetrad中Na+(2)沿α方向移動
Na+(2)沿α方向的移動以X=0.1750 nm(X為Na+(2)距平面I的距離)為對稱軸.Na+(2)沿α方向移動是指:第1片G-tetrad的坐標不變,改變Na+(2)距平面I的距離,如圖3(a)所示.首先將Na+(2)沿α正方向,每隔0.0100 nm取一個結構;再將Na+(2)沿α負方向每隔0.0100 nm取一個結構,到0.3250 nm后每隔0.1000 nm取一個結構,到1.6250 nm后每隔0.5000 nm取一個結構,直到4.0250 nm.圖3(b)展示了MP2水平和ABEEMσπ/MM模型下,Na+(2)沿α方向移動時,Na+(2)-G-tetrad體系結合能變化曲線.

圖3 Na+(2)沿α方向移動的正視圖(a)和結合能變化曲線(b) Fig.3 The front view(a)and the binding energy curve(b)of Na+(2)moving along the α orientationd:the distance between Na+and its equilibrium position along corresponding moving direction;The data in parentheses denote the coordinate ofextremal point for Na+(2)-G-tetrad when Na+(2)moves along the α orientation obtained from ABEEMσπ/MM model;the view of big pane is amplificatory view of small pane.

圖4 Na+(2)沿β方向移動的俯視圖(a)和結合能變化曲線(b)Fig.4 The vertical view(a)and the binding energy curve(b)of Na+(2)moving along the β orientationThe data in parentheses denote the coordinates of extremal points for Na+(2)-G-tetrad when Na+(2)moves along the β orientation.
根據總的結合能變化曲線可以看出,Na+(2)與平面I之間距離為0.1000 nm時(圖3中A點位置),復合物Na+(2)-G-tetrad的結合能最大,結構最穩定.而實驗構型中K+在平面I內時,結構最穩定.隨著Na+(2)向α正方向或α負方向移動,該復合物的結合能逐漸減小,直到距離小于-1.8500 nm或大于2.0500 nm后,結合能約等于0.00 kJ·mol-1,說明Na+(2)與G-tetrad完全分開,不存在相互作用,相當于兩個獨立的個體.
2.2.2 Na+(2)-G-tetrad中Na+(2)沿β方向移動
由于Na+(2)沿β正負方向的移動是對稱的,所以本文僅以向β正方向移動為例進行詳細分析.Na+(2)沿β方向移動指:第1片G-tetrad的坐標不變,只是沿第1片G-tetrad中兩鳥嘌呤的夾縫在平面I中的投影方向移動,改變Na+(2)到平衡位置的距離,如圖4(a)所示.首先將Na+(2)沿β正方向每隔0.0200 nm取一個結構,到0.5150 nm后每隔0.0500 nm取一個結構,到1.5650 nm后每隔0.5000 nm取一個結構,直到4.6650 nm.圖4(b)展示了MP2水平和ABEEMσπ/ MM模型下,Na+(2)沿β方向移動時,Na+(2)-G-tetrad體系的結合能變化曲線.
可以看出平衡位置時(圖4中A點位置)結合能最大,此時體系最穩定.隨著Na+(2)向β正方向移動,到距離平衡位置0.2350 nm時(圖4中B點位置)體系到達結合能的一個局部極小值點,Na+(2)與右側氧原子的作用削弱了原有的O…H—N氫鍵,相應降低了體系的穩定性;隨著Na+(2)繼續向β正方向移動,到距離平衡位置0.6150 nm時(圖4中C點位置)體系到達結合能的一個局部極大值點;Na+(2)繼續向β正方向移動,結合能下降,直到Na+(2)移動到距平衡位置3.1650 nm后結合能約等于0.00 kJ·mol-1, Na+(2)與G-tetrad完全分開.
2.2.3 Na+(2)-G-tetrad中Na+(2)沿γ方向移動

圖5 Na+(2)沿γ方向移動的俯視圖(a)和結合能變化曲線(b)Fig.5 The vertical view(a)and the binding energy curve(b)of Na+(2)moving along the γ orientationThe data in parentheses denote the coordinates of extremal points for Na+(2)-G-tetrad when Na+(2)moves along the γ orientation.
同樣Na+(2)沿γ正負方向的移動也是對稱的,以下以γ正方向移動為例進行詳細分析.Na+(2)沿γ方向移動指:第1片G-tetrad的坐標不變,只是沿第1片G-tetrad中兩鳥嘌呤的對角線在平面I中的投影方向移動,改變Na+(2)到平衡位置的距離,如圖5(a)所示.首先將Na+(2)沿γ正方向每隔0.0200 nm取一個結構,到0.8200 nm后每隔0.2000 nm取一個結構,到1.8200 nm后每隔0.5000 nm取一個結構,直到3.8200 nm.圖5(b)展示了MP2水平和ABEEMσπ/MM模型下,Na+(2)沿γ方向移動時,Na+(2)-G-tetrad的結合能變化曲線.
與Na+(2)沿β方向移動一致,仍然是平衡位置時(圖5中A點位置)結合能最大,體系最穩定.隨著Na+(2)向γ正方向移動,體系穩定性降低.到距離平衡位置0.3600和0.5800 nm時(圖5中B點和D點位置)體系到達結合能的局部極小值點,到距離平衡位置0.4600和0.8000 nm時(圖5中C點和E點位置)體系到達結合能的局部極大值點,直到Na+(2)距離平衡位置2.3200 nm后結合能約等于0.00 kJ· mol-1,Na+(2)與G-tetrad完全分開.
對于 Na+(2)的這三條移動路線,MP2和ABEEMσπ/MM方法計算的結合能的絕對平均偏差分別為7.36、6.07和12.30 kJ·mol-1,表明ABEEMσπ/ MM方法計算的結合能與MP2方法得到的結果有很好的一致性.
2.3 Na+和兩片G-tetrad作用的結合能
以上結果說明ABEEMσπ/MM模型能很好地模擬MP2水平上,一個Na+在一片G-tetrad中的移動路線.由于受計算機的限制,從頭算很難模擬兩片甚至更大的體系.以下應用ABEEMσπ/MM模型預測了G-四鏈體中Na+的移動路線.

圖6 Na+(2)-G-tetrads中Na+(2)分別沿α(a)、β(b)、γ(c)三個方向移動和Na+(3)-G-tetrads中Na+(3)分別沿α(a′)、β′(b′)、γ′(c′)三個方向移動時的結合能變化曲線Fig.6 Binding energy curves of Na+(2)-G-tetrads for Na+(2)moving along the α(a),β(b),γ(c)orientations and Na+(3)-G-tetrads for Na+(3)moving along the α(a′),β′(b′),γ′(c′)orientationsThe data in parentheses denote the coordinates of extremal points along the corresponding orientation.
分別取第1和第2片G-tetrad和其中的Na+(2) (Na+(2)-G-tetrads),以及第2和第3片G-tetrad和其中的Na+(3)(Na+(3)-G-tetrads)作為平衡結構(圖2).對于Na+(2)-G-tetrads結構,Na+(2)同樣沿α、β、γ三個方向移動.對于Na+(3)-G-tetrads結構,通過Na+(3),且垂直于α方向取一個平面II,將第2片G-tetrad中兩鳥嘌呤的夾縫方向在平面II中的投影定義為β′方向;將第2片G-tetrad中兩鳥嘌呤的對角線方向在平面II中的投影定義為γ′方向.以Na+(3)所在位置(平衡位置)為起點,討論了該離子分別沿α、β′、γ′三個方向的移動.由于沿各條路線移動時,正負方向的移動是對稱的,所以僅以向正方向移動為例進行詳細分析.通過比較各條移動路線中,Na+與兩片G-tetrad作用的結合能,來判斷Na+的移動路線.結合能計算公式如下:

其中E[Na+]為Na+的能量,E[G-tetrads]為兩片G-tetrads復合物的能量,E[Na+-G-tetrads]為Na+-G-tetrads復合物的能量.
圖6展示了ABEEMσπ/MM模型下,Na+(2)和Na+(3)沿各條路線移動時,Na+-G-tetrads體系的結合能變化曲線.從圖中可以看出,對于α方向,Na+(2)-G-tetrads和Na+(3)-G-tetrads中的Na+都是處于平衡位置(圖6中A點)時,體系的結合能最大,結構最穩定.Na+(2)和Na+(3)分別沿α方向移動時,分別需克服564.63和562.10 kJ·mol-1的結合能移出G-四鏈體.對于β、γ、β′和γ′四個方向,圖6各圖中B點和D點位置是體系結合能的局部極小值點;C點位置是體系結合能的局部極大值點.其中圖6(b)中D點代表Na+(2)與G-tetrads中的N42距離(0.1680 nm)較近,vdW排斥作用(-2856.41 kJ·mol-1)較大;圖6 (c)、6(b′)、6(c′)中B點分別代表Na+(2)或Na+(3)與G-tetrads中的O61、N40、O39距離(0.1510、0.1480、0.1190 nm)較近,vdW排斥作用(-1775.25、-2356.86、-31836.73 kJ·mol-1)較大.理論上,存在這么大的排斥能Na+很難從β、γ、β′和γ′四個方向移出.綜上所述,可以預測G-四鏈體中的三個Na+最有可能沿α方向依次移出.
3 結論
Na+-G-四鏈體是一個明顯的極化體系,而ABEEMσπ/MM模型較其它力場能更好地反映體系的極化現象,因此應用ABEEMσπ/MM模型對此體系進行了研究.ABEEMσπ/MM模型驗證了:Na+-G-tetrad結構采用平行型的氫鍵,而Na+不存在時, G-tetrad的四個鳥嘌呤通過交叉型氫鍵存在.Na+的存在改變了G-tetrad的氫鍵方式.通過研究一片G-tetrad和一個Na+的相互作用表明:Na+沿α方向移動與平面I的距離為0.1000 nm時,體系最穩定,而實驗構型中的K+在平面I內時體系最穩定.并且該方法計算的Na+(2)-G-tetrad體系中Na+(2)分別沿α、β、γ三個方向移動的結果與從頭算(MP2)的結果有很好的一致性,充分驗證了模型的準確性.在此基礎上,應用ABEEMσπ/MM模型研究了Na+(2)-G-tetrads和Na+(3)-G-tetrads中兩個Na+的移動路線.由于兩個Na+沿β、γ、β′和γ′方向移動時,與兩片G-tetrad之間存在很大的排斥能,因此無法從這四個方向移出.而這兩個Na+沿α方向移動,需克服的結合能相對較小,由此可以預測,G-四鏈體中的三個Na+最有可能沿α方向依次移出.本文對Na+-G-tetrad和Na+-G-tetrads體系的研究和探討,為進一步應用ABEEMσπ/ MM模型進行G-四鏈體中離子交換通道的動力學模擬打下堅實的基礎.
致謝: 感謝Jay William Ponder教授(Department of Biochemistry&Molecular Biophysics,School of Medicine,Washington University)提供Tinker程序.
1 Han,H.Y.;Hurley,L.H.Trends Pharm.Sci.,2000,21:136
2 Sket,P.;Cmugelj,M.;Plavec,J.Bioorg.Med.Chem.,2004,12: 5735
3 Davis,J.T.Angew.Chem.Int.Edit.,2004,43:668
4 Shafer,R.H.;Smirnov,I.Biopolymers,2000,56:209
5 Moine,H.;Mandel,J.L.Science,2001,294:2487
6 Arthanari,H.;Bolton,P.H.Chem.Biol.,2001,8:221
7 Lane,A.N.;Jenkins,T.C.Curr.Org.Chem.,2001,5:845
8 Pennarun,G.;Granotier,C.;Gauthier,L.R.Oncogene,2005,24: 2917
9 Burger,A.M.;Dai,F.;Schultes,C.M.Cancer Res.,2005,65: 1489
10 Tuntiwechapikul,W.;Lee,J.T.;Salazar,M.J.Am.Chem.Soc., 2001,123:5606
11 Cuesta,J.;Read,M.A.;Neidle,S.Min.Rev.Med.Chem.,2003,3: 11
12 Kerwin,S.M.Curr.Pharm.Des.,2000,6:441
13 Neidle,S.;Read,M.A.Biopolymers,2000,56:195
14 Perry,P.J.;Arnold,J.R.P.;Jenkins,T.C.Expert.Opin Investig. Drugs,2001,10:2141
15 Alberti,P.;Lacroix,L.;Guittat,L.;Helene,C.;Mergny,J.L.Min. Rev.Med.Chem.,2003,3:23
16 Hud,N.V.;Smith,F.W.;Anet,F.A.;Feigon,J.Biochemistry, 1996,35:15383
17 Qin,Y.;Hurley,L.H.Biochimie,2008,90:1149
18 Shen,X.Y.;Lü,Y.;Li,S.M.Acta Phys.-Chim.Sin.,2009,25: 783 [沈新媛,呂 洋,李慎敏.物理化學學報,2009,25:783]
19 Meng,F.C.;Xu,W.R.;Liu,C.B.Chem.Phys.Lett.,2004,389: 421
20 Gu,J.;Leszczynski,J.J.Phys.Chem.A,2000,104:6308
21 Ma,L.;Iezzi,M.;Kaucher,M.S.;Lam,Y.F.;Davis,J.T.J.Am. Chem.Soc.,2006,128:15269
22 Tsuzuki,S.;Uchimaru,T.;Matsumura,K.;Mikami,M.;Tanabe,K. J.Chem.Phys.,1999,110:11906
23 Yang,Z.Z.;Wu,Y.;Zhao,D.X.J.Chem.Phys.,2004,120:2541 24 Wu,Y.;Yang,Z.Z.J.Phys.Chem.A,2004,108:7563
25 Yang,Z.Z.;Liu,Y.J.Acta Phys.-Chim.Sin.,2009,25:928 [楊忠志,劉永軍.物理化學學報,2009,25:928]
26 Li,X.;Yang,Z.Z.J.Phys.Chem.A,2005,109:4102
27 Qian,P.;Yang,Z.Z.Acta Phys.-Chim.Sin.,2006,22:561 [錢 萍,楊忠志.物理化學學報,2006,22:561]
28 Liu,C.;Yang,Z.Z.Sci.China.Ser.B-Chem.,2009,38:1461 [劉 翠,楊忠志.中國科學B輯:化學,2009,38:1461]
29 Wang,F.F.;Gong,L.D.;Zhao,D.X.J.Mol.Struct.-Theochem, 2009,909:49
30 Li,X.;Yang,Z.Z.J.Chem.Phys.,2005,122:084514
31 Pople,J.A.;Head-Gordon,M.;Raghavachari,K.J.Chem.Phys., 1987,87:5968
32 Parkinson,G.N.;Lee,M.P.H.;Neidle,S.Nature,2002,417:876
September 11,2009;Revised:November 20,2009;Published on Web:December 28,2009.
Mobility of Na+in a G-Quadruplex
GUO Ci LIU Cui YANG Zhong-Zhi*
(School of Chemistry and Chemical Engineering,Liaoning Normal University,Dalian 116029,Liaoning Province,P.R.China)
The Na+-G-quadruplex complex is a polarized system and the mobility of Na+during its formation or decomposition is still unclear.The atom bond electronegativity equalization method at the σπ level fused into molecular mechanics(ABEEMσπ/MM)model clearly defines the lone-pair electron,σ bond and π bond sites in addition to the atomic sites.The partial charge fluctuation is calculated in accordance with a change in the molecular environment and so this method should account well for the polarization effect.In this paper,we discuss some properties for the Na+-G-tetrad complex including its geometry,charge distribution,and binding energy according to the ABEEMσπ/MM method.We also investigate these properties for the Na+-G-tetrad complex using the ab initio method at the MP2/6-31G(d,p)level.The ABEEMσπ/MM results are in good agreement with the ab initio results.The presence of Na+changes the hydrogen bonds in the G-tetrad.By comparing the binding energy of the system for every Na+mobile path, we predict that the most probable path is that three Na+ions move away individually from the G-quadruplex along the α orientation.This study lays a solid foundation for the dynamic simulation of ion exchange channels in a G-quadruplex using the ABEEMσπ/MM model.
G-quadruplex;Na+-G-tetrad;ABEEMσπ/MM method;Ab initio;Mobile path
O641
*Corresponding author.Email:zzyang@lnnu.edu.cn.Tel:+86-411-82159607.
The project was supported by the National Natural Science Foundation of China(20633050,20873055)and Foundation of Department of Education of Liaoning Province,China(2008S133,2009T057,LNET RC0503).
國家自然科學基金(20633050,20873055)和遼寧省教育廳基金(2008S133,2009T057,LNET RC0503)資助項目
猜你喜歡
計算機應用(2022年2期)2022-03-01 12:33:42
哲學評論(2021年2期)2021-08-22 01:53:34
計算機應用(2021年4期)2021-04-20 14:06:36
計算機應用(2021年1期)2021-01-21 03:22:38
新世紀智能(數學備考)(2020年11期)2021-01-04 00:38:16
中華詩詞(2019年7期)2019-11-25 01:43:04
影視與戲劇評論(2016年0期)2016-11-23 05:26:01
現代企業(2015年9期)2015-02-28 18:56:50
小天使·一年級語數英綜合(2015年2期)2015-01-14 06:35:05
新高考·高一物理(2014年1期)2014-09-18 01:26:07
