新致病基因在顱縫早閉Crouzon綜合征中的初步機制研究
2012-01-16 03:16:22楊嫻嫻JodieHatfieldSusanHinze穆雄錚PeterAndersonBarryPowell
組織工程與重建外科雜志 2012年5期
關鍵詞:小鼠
楊嫻嫻 Jodie T Hatfield Susan J Hinze 穆雄錚 Peter J Anderson Barry C Powell
新致病基因在顱縫早閉Crouzon綜合征中的初步機制研究
楊嫻嫻 Jodie T Hatfield Susan J Hinze 穆雄錚 Peter J Anderson Barry C Powell
目的 研究 Rbp4(Retinol binding protein 4)、Gpc3(Glypican family of growth factor binding protein 3)、C1qtnf3(Collagenous repeat-containing sequence of 26 KDa protein)等新致病基因在顱縫早閉癥中的發病機制,為疾病非手術治療奠定理論基礎。方法 以Crouzon綜合征Fgfr2cC342Y/+小鼠為實驗模型,應用MicroCT和組織學染色,研究小鼠顱縫閉合模式;以Fgfr2cC342Y/+模型,RT-qPCR研究Rbp4、Gpc3、C1qtnf3等新致病基因的表達差異,初步探討其在顱縫閉合過程中的調控作用;以體外培養的Fgfr2cC342Y/+小鼠顱縫細胞為模型,研究基因突變動物細胞增殖與代謝改變。結果 獲得Fgfr2cC342Y/+小鼠后額縫、冠狀縫、人字縫、矢狀縫等顱縫閉合模式,隨顱骨發育、顱縫閉合,OC(Osteocalcin)、ALP(Alkaline phosphatase)表達增加,目的基因 Rbp4、Gpc3、C1qtnf3 表達下降,Msx2(Muscle segment homeobox gene 2)表達增加,雜合子與野生型小鼠之間均存在顯著統計學差異,與前期人顱縫組織Microarray研究結果一致。Gpc1(Glypican family of growth factor binding protein 1)、FliI(Flightless I)在顱縫閉合中的表達較恒定,野生型與雜合子之間未見明顯統計學差異。體外培養Fgfr2cC342Y/+小鼠顱縫細胞,CellTiter96 MTS和Quant-iT Picogreen dsDNA細胞增殖與代謝分析結果顯示,Fgfr2功能獲得性突變可促進冠狀縫細胞的增殖,從而導致成骨增加,顱縫早閉。結論 Fgfr2cC342Y/+模型新致病基因表達趨勢與前期人顱縫組織Microarray研究結果一致,Rbp4、Gpc3、C1qtnf3可能在顱縫早閉中具重要調控作用。
顱縫早閉癥 Fgfr2cC342Y/+小鼠 Crouzon綜合征 RT-qPCR 微量CT Rbp4 Gpc3 C1qtnf3
顱縫早閉癥是一種常見的先天性顱頜面畸形,新生兒中發病率約為1/2 500,僅次于唇腭裂畸形。由于顱縫提早發生骨性閉合,導致顱腔狹小、顱面骨畸形、顱內壓增高等復雜的顱頜面畸形綜合征,如Crouzon綜合征、Apert綜合征等。嚴重者可引起視力減退甚至失明、腦發育受阻及智力發育障礙,嚴重影響了患兒的正常發育、生理功能,對患兒的社會生活和心理健康產生了諸多的負面效應,甚至可在發育過程中因危險的并發癥而危急生命[1]。目前,對該癥唯一的治療手段為出生后經顱徑路畸形矯正、顱腔減壓,多需2~3次分期手術,手術風險大,并發癥多,費用高,且術后效果不理想。因此,對這類疾病遺傳機制的研究與防治具有重要的社會意義。
Coussens等[2]通過5名顱縫早閉癥患兒(非綜合征性顱縫早閉癥4人,Apert綜合征1人)取材的16條顱縫組織(含受累及正常顱縫),應用Microarray技術,比較了完全閉合、活躍閉合、未閉合3種顱縫間18 000個基因表達趨勢,除 FGFR(Fibroblast growth factor receptor)1~3外,還包括一系列與顱骨發育、顱縫閉合(成骨分化、轉錄、信號轉導、骨塑形、細胞周期和細胞凋亡)相關的新基因。結果顯示,受累顱縫與正常顱縫相比,視黃醇結合蛋白-4(Retinol binding protein 4,Rbp4)表達下調 37倍、磷脂酰肌醇蛋白聚糖-3(Glypican family of growth factor binding protein 3,Gpc3)表達下調 7倍、C1Q 腫瘤壞死因子相關蛋白-3(Collagenous repeat-containing sequence of 26 KDa protein,C1qtnf3) 表達下調 20倍,提示Rbp4、Gpc3、C1qtnf3等新基因在顱骨發育、顱縫閉合中可能存在調控作用。
由于倫理道德問題及復雜實驗技術的挑戰,從患者或正常人群中獲取顱縫組織、細胞用于大規模的實驗研究相當困難。因此,制造相關致病基因修飾小鼠模型,進行顱縫早閉癥遺傳分子機制及基因或細胞治療的研究是可行的途徑。FGFR突變可導致Apert、Pfeiffer和Crouzon等顱縫早閉綜合征,現有的基因修飾小鼠模型包括FGFR2+/S252W、FGFR2+/C342Y和 FGFR1+/P250Arg等[3-6]。 Eswarakumar等[5]應用小鼠基因打靶技術,定點突變小鼠胚胎干細胞Fgfr2c基因,以半胱氨酸代替酪氨酸位點(Cys342Tyr),成功制造出FGFR2功能獲得性突變 Fgfr2cC342Y/+模型。Fgfr2cC342Y/C342Y純合子小鼠表現出多關節融合、腭裂和肺氣管畸形等,于出生后不久死亡;Fgfr2cC342Y/+雜合子小鼠可存活并繁殖,顯示出Crouzon綜合征表現型。
本實驗將以Fgfr2cC342Y/+小鼠為模型,分析上述新基因在閉合與未閉合顱縫間的表達差異,驗證前期的人Microarray研究結果,初步探討基因在顱縫閉合過程中的調控作用。
1 材料與方法
1.1 實驗動物
實驗采用Fgfr2cC342Y/+小鼠。Fgfr2cC342Y/+雄性小鼠(美國華盛頓大學Chad Perlyn教授惠贈),與Swiss雌性小鼠(購自阿德萊德婦女兒童醫院動物實驗中心)配種繁殖。子代中將出現野生型(WT,+/+)或雜合子(Het,+/-)2種基因型。提取鼠尾組織抽提基因組DNA進行基因型鑒定,并進行鼠耳標記。孕鼠經剖宮獲取胎鼠,切取實驗用顱縫組織后,提取鼠尾組織進行基因型鑒定。按“QIAGEN全血和組織DNA抽提試劑盒”進行操作,PCR擴增突變基因外顯子,引物序列為5'-CAAGCAAGCTCAACAG GAGAG-3'(Forward) 和 5'-GCTGTGCTGCTGAGAGTTTTG-3'(Reverse)。PCR產物瓊脂糖凝膠電泳分析,根據野生型出現224 bp條帶,雜合子出現290 bp突變等位基因條帶,確定基因型。
1.2 主要試劑
Real-time PCR引物(Geneworks公司,澳大利亞);TURBO DNA-freeTM試劑盒、BigDye Terminator V3.1測序試劑盒、AmpliTaqGold DNA polymerase(Applied Biosystems公司,美國);PCR產物純化試劑盒、Glycogen(Roche 公司,瑞士);RNase ZAP,DEPC,青鏈霉素,Alizarin Red S、Tris(Sigma-Aldrich 公司,美國);Trizol、SuperScriptTMⅢ逆轉錄試劑盒、FBS、α-MEM 培養液、10mM dNTPs(Invitrogen 公司,美國);氯仿、異丙醇、EDTA(APS Ajax Finechem 公司,澳大利亞);100× SYBR green(Abgene公司,英國);KAPA SYBR FAST qPCR 試劑盒(Kapa Biosystems公司,美國);geNormTMHousekeeping Gene Selection試劑盒(Primer Design公司,英國);Lillie Mayer(Sigma Diagnostics公司,美國);eosin Y(Surgipath公司,美國);其余試劑均為分析純。
1.3 主要儀器與設備
GeneAmp PCR System 9700基因擴增儀(PE Applied Biosystems公司,美國);Rotor-Gene 6000 Real-Time PCR 儀(Corbett Reserach,Qiagen公司,德國);100 孔板(Corbett Research 公司,澳大利亞);高速臺式離心機5415D(Eppendorf公司,德國);G:BOX凝膠成像分析儀(Syngene公司,英國);Skyscan 1072/1076 MicroCT掃描儀(Skyscan公司,比利時);NanoVue超微量分光光度計(GE公司,美國);倒置相差顯微鏡(Leica公司,德國)。
1.4 方法
1.4.1 Fgfr2cC342Y/+小鼠顱縫閉合模式研究
1.4.1.1 MicroCT掃描
選擇胚胎 16.5 d、18.5 d 及出生 0 d、1 d、5 d、10 d的Fgfr2cC342Y/+小鼠(n=5),解剖獲取完整顱蓋骨(自鼻根部至頸根部,兩側眼耳平面,包括骨膜、顱骨及其下硬腦膜),分管標記置于10%中性福爾馬林瓶中,置于Skyscan MicroCT 1072儀器中掃描,三維圖像重建分別由NRecon、CTan v.1.5.0.2和ANT軟件完成。
1.4.1.2 HE染色
完成MicroCT掃描后,分別切取冠狀縫、后額縫、矢狀縫、人字縫復合組織(包括骨膜、顱縫間質、成骨緣及其下硬腦膜),按不同天數及不同顱縫分管標記,5 d以上標本5%EDTA脫鈣1~3 d,經組織脫水、二甲苯透明、浸蠟、包埋常規處理,進行HE染色,顯微鏡下觀察不同顱縫不同天數的閉合模式。
1.4.2 Fgfr2cC342Y/+小鼠新致病基因表達研究
1.4.2.1 顱縫標本制備
選擇出生后 0 d、1 d、5 d、10 d 的 Fgfr2cC342Y/+小鼠(n=6),分別切取冠狀縫、后額縫、矢狀縫、人字縫復合組織和顱頂骨(顱縫復合組織包括顱縫間質、成骨緣及其下硬腦膜,切取顱縫時盡可能留小于1 mm成骨緣),分管標記置于5 mL EP管中,-80℃保存。
1.4.2.2 RNA抽提與逆轉錄
顱縫組織按Trizol法抽提總RNA,加入糖原充分混勻共沉淀,應用20 μL DEPC蒸餾水55℃促溶10 min獲得RNA標本。A260/A280、1%瓊脂糖凝膠電泳進行RNA純度及濃度測定。用Ambion公司TURBO DNA-freeTM試劑盒進行DNase處理,去除RNA樣品中的DNA污染。
查詢NCBI Genbank獲取小鼠目的基因編碼序列,引物由Primer Premier 5.0軟件設計(表1),由澳大利亞Geneworks公司合成,引物干粉按產品使用說明稀釋至原液,濃度為100 pmol/μL,工作液再稀釋 25 倍為 4 pmol/μL,測序濃度為 3.2 pmol/μL。
將抽提的RNA溶液稀釋,取100 ng(至少2 μL),加 DEPC水至 8 μL,采用 SuperScriptTMⅢ First-Strand Synthesis System試劑盒20 μL體系進行逆轉錄反應。cDNA經不含RNase/DNase的高壓水1∶3比例稀釋。所有反應均設定相應No-RT陰性對照(即逆轉錄反應中,由高壓水替代SuperscriptⅢ逆轉錄酶)。
1.4.2.3 RT-qPCR
驗證目的基因與內參基因的擴增效率均為(100±5)%。RT-qPCR 采用 KAPA SYBR FAST qPCR Kit試劑盒10 μL qPCR反應體系,在Rotor-Gene 6000 Real-time PCR儀上進行PCR反應,按①95℃3 min,②95 ℃ 3 sec,③60 ℃ 25 sec,回到步驟②,共40個循環。每一個RNA標本均做2副孔,每一板PCR反應均設定空白對照(NTCs),反應結束后通過熔解曲線分析、凝膠電泳以及測序等鑒定產物的特異性。
針對Fgfr2cC342Y/+小鼠顱縫組織,采用小鼠geNormTMHousekeeping Gene Selection試劑盒(提供候選內參基因PCR擴增引物)進行geNorm分析,顯示Cyc1-Gapdh-Canx為最佳組合,選取這3個管家基因Ct值的幾何平均值作為內參來對目的基因表達進行校正。
1.4.2.4 目的基因RT-qPCR數據分析
Ct數據分析分別采用傳統 2-ΔΔCt法[7]及 qbasePLUS軟件法[8-9],Microsoft Excel、Graphpad Prism 5 軟件行Student TTEST檢驗(雙尾、雙樣本等方差假設)統計,P<0.05則認為兩者具顯著性差異。
1.4.3 細胞增殖與代謝測定
1.4.3.1 細胞培養
選擇出生后3 d的Fgfr2cC342Y/+小鼠6只(野生型、突變型各3只),獲取各顱縫及非顱縫區頂骨(剝離硬腦膜和骨膜),分別接種于24孔培養板底部,略微干燥后每孔加入600 mL培養液,37℃和5%CO2的培養箱中培養。第3天換液,觀察細胞生長情況,接著每3天換1次液。待細胞長滿培養孔,0.25%的胰酶消化傳代,轉移至12孔板,繼續培養,二次傳代至6孔板,以獲得足夠細胞數,三次傳代至96孔板以進行MTS和dsDNA實驗。
1.4.3.2 CellTiter96 AQueous One Cell Proliferation Assay
將培養在6孔板的冠狀縫細胞和頂骨細胞,應用胰酶消化計數,傳代至96孔板培養,每孔含8 000細胞數、0.2 mL培養液,每一只動物來源的細胞作6個副孔。傳代2板,一板為傳代當日4 h后細胞貼壁,一板為培養3 d后,記Plate-1,2,均應用CellTiter96 AQueous One Cell Proliferation Assay Kit來測定細胞增殖代謝的情況。吸除培養基,每孔加入0.1 mL 新鮮培養基,20 μL CellTiter96 MTS solution,37 ℃ 、5%CO2培養4 h,每隔30 min,以ELISA-plate reader測其OD 490 nm吸光值。
1.4.3.3 Quant-iTTMPicoGreen dsDNA Cell Proliferation Assay
將培養在6孔板的冠狀縫細胞和頂骨細胞,應用胰酶消化計數,傳代至96孔板培養,每孔含8 000細胞數、0.2 mL培養液,每一只動物來源的細胞作6個副孔。傳代2板,一板為傳代當日4 h后細胞貼壁,一板為培養3 d后,記Plate-3,4,均應用QuantiTTMPicoGreen dsDNA Reagent and Kits,定量測定dsDNA合成,分析細胞增殖情況,同時觀察Plate-1,2 CellTiter96檢測后細胞的增殖情況,探討MTS Kit對細胞的毒性作用。
吸除MTS培養基,PBS洗3次,每孔加入10 μL蛋白酶 K、80 μL PBS、10 μL 甘氨酸(1 M 甘氨酸DEPC、pH=8.0)消化細胞,混勻后孵育 1 h,每孔加入 100 μL PicoGreen Assay work solution,每板按說明構建DNA Standard Curve,所有標本室溫避光孵育2~5 min,以FLUOstar OPTIMA微孔板檢測儀,熒光激發波長480 nm、發射波長520 nm,測其吸光值。
2 結果
2.1 Fgfr2cC342Y/+小鼠顱縫閉合模式
2.1.1 雜合子小鼠冠狀縫
MicroCT及HE染色結果均顯示,Fgfr2cC342Y/+小鼠野生型冠狀縫在所有取材時間點,直至出生后第10天仍保持開放,而Fgfr2cC342Y/+雜合子冠狀縫于胚胎18.5 d開始閉合,第0、1、5天持續閉合,出生后第 10天完全閉合(圖1,2)。
2.1.2 野生型及雜合子小鼠后額縫
Fgfr2cC342Y/+野生型及雜合子小鼠出生后第5天,后額縫仍開放,至第10天,少數后額縫出現散在的閉合位點,并非以自前向后的模式閉合,顱縫內層骨面(硬腦膜面)較外層骨面(骨膜面)融合早(圖3)。
2.1.3 矢狀縫、人字縫
MicroCT及HE染色結果均顯示,Fgfr2cC342Y/+小鼠野生型或雜合子,于出生后第10天,矢狀縫、人字縫均保持開放。
2.2 Fgfr2cC342Y/+小鼠目的基因表達
隨顱骨發育、顱縫閉合,野生型和雜合子小鼠冠狀縫OC表達均增加,但兩者之間無顯著性差異。ALP表達隨冠狀縫閉合逐漸增加,出生后5 d、10 d,雜合子小鼠ALP表達水平較野生型顯著增加(圖4)。
目的基因 Rbp4、Gpc3、C1qtnf3 表達下降,Msx2表達增加,雜合子與野生型小鼠之間均存在顯著差異,與前期人顱縫組織Microarray研究結果一致。Gpc1、FliI在顱縫閉合中的表達較恒定,野生型與雜合子之間未見明顯差異。
2.3 Fgfr2cC342Y/+小鼠顱縫細胞增殖與代謝
MTS和PicoGreen結果ANOVA統計均顯示,體外培養的Fgfr2cC342Y/+小鼠非顱縫區頂骨細胞(PB)和冠狀縫細胞(Cor)增殖速度存在差異,突變型頂骨細胞較野生型增殖略快,但無明顯差異;而突變型冠狀縫細胞較野生型增殖顯著增加(圖5)。
通過PicoGreen測定MTS分析后細胞(Plate-1,2)與新鮮細胞(Plate-3,4)間的增殖差異,結果表明,經MTS分析后,培養板中的細胞數較新鮮細胞減少約1/3~1/2。盡管如此,應用MTS分析后細胞進行PicoGreen分析,仍可出現冠狀縫突變型較野生型增殖增加,并具統計學差異。
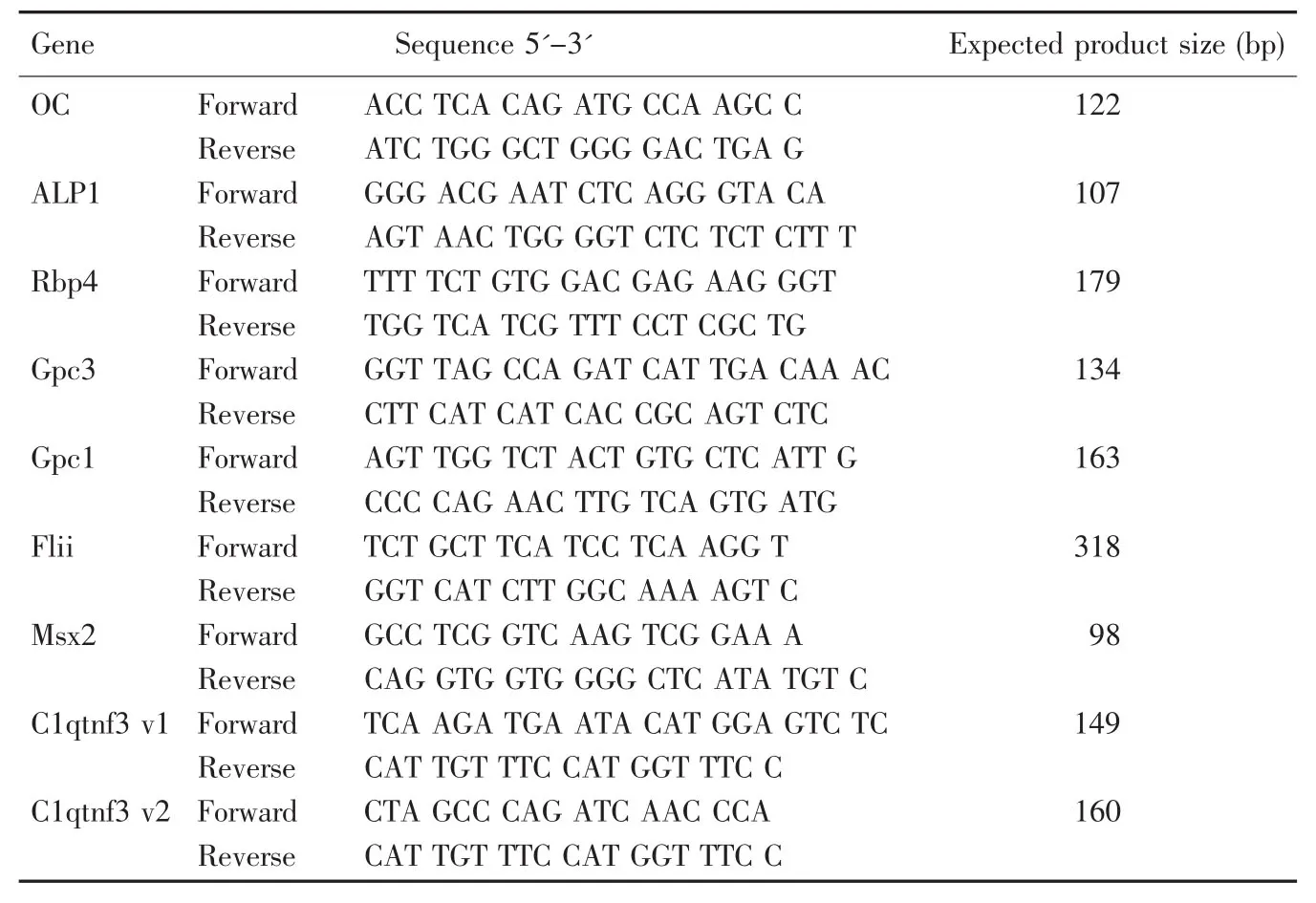
表1 定量PCR檢測基因及引物序列Table 1 RT-qPCR target gene and primer list
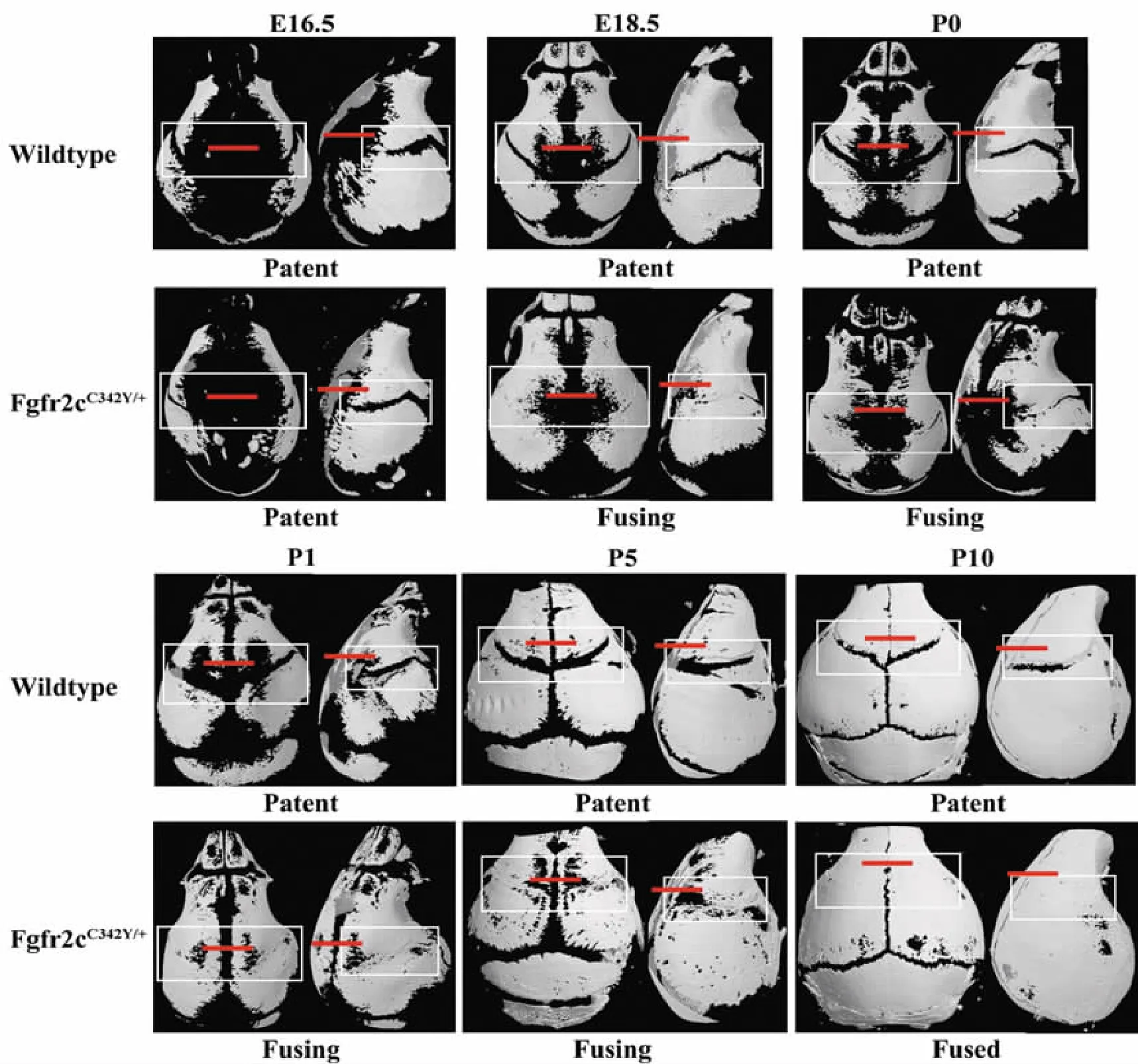
圖1 Fgfr2cC342Y/+小鼠冠狀縫閉合MicroCT檢測Fig.1 Representative MicroCT images comparing wildtype and Fgfr2cC342Y/+mice sutures
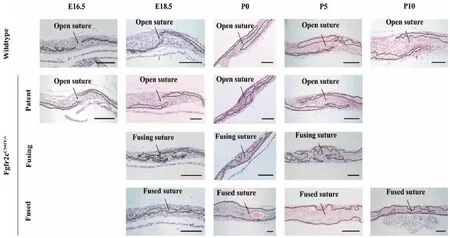
圖2 Fgfr2cC342Y/+小鼠冠狀縫閉合HE染色Fig.2 HE staining of wildtype and Fgfr2cC342Y/+mice coronal sutures

圖3 Fgfr2cC342Y/+小鼠后額縫閉合MicroCT和HE染色Fig 3 Representative images displaying posterior frontal suture fusion in Fgfr2cC342Y/+mice
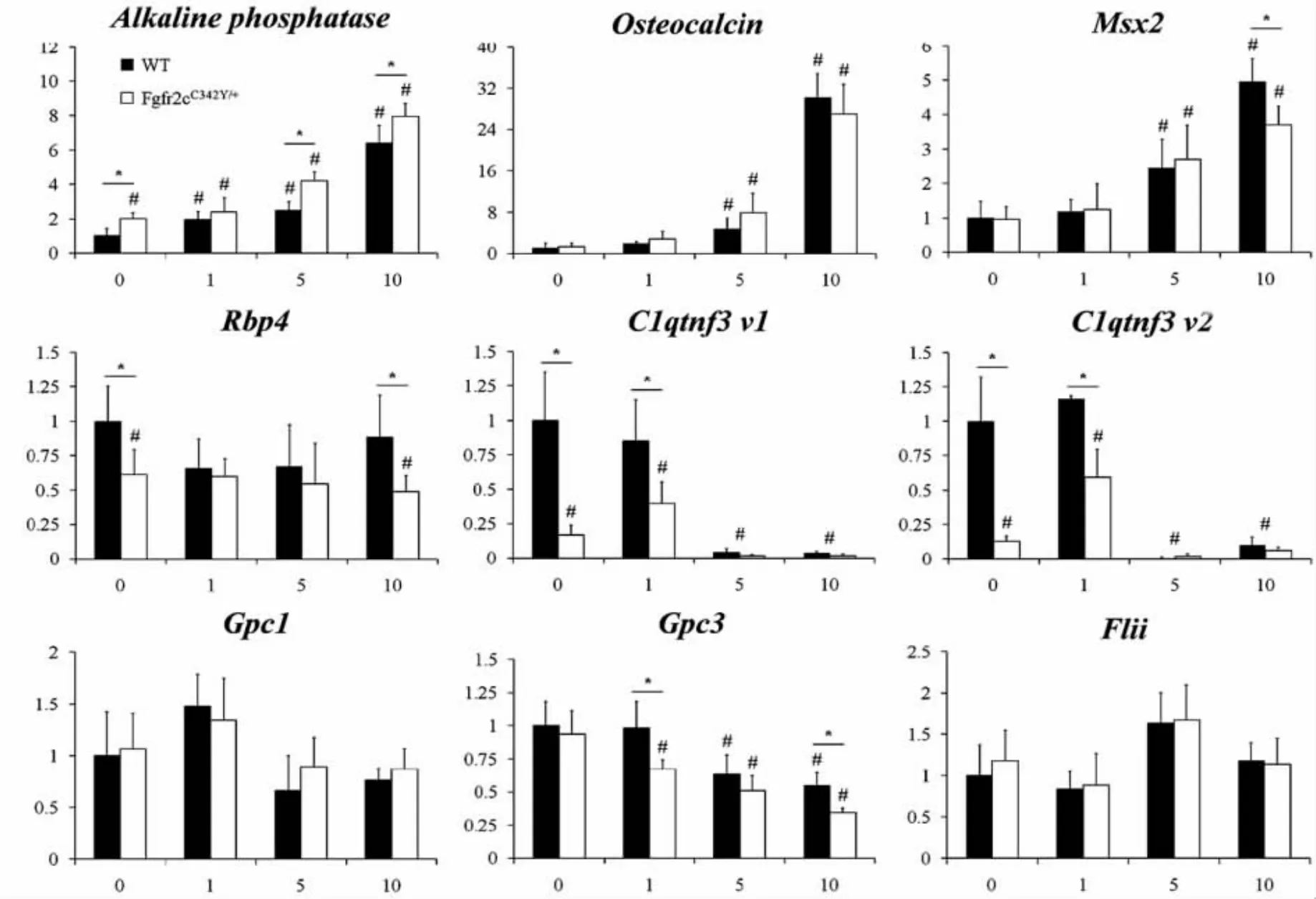
圖4 Fgfr2cC342Y/+小鼠冠狀縫目的基因表達相對定量圖Fig.4 Relative target gene expression from wildtype and Fgfr2cC342Y/+coronal sutures
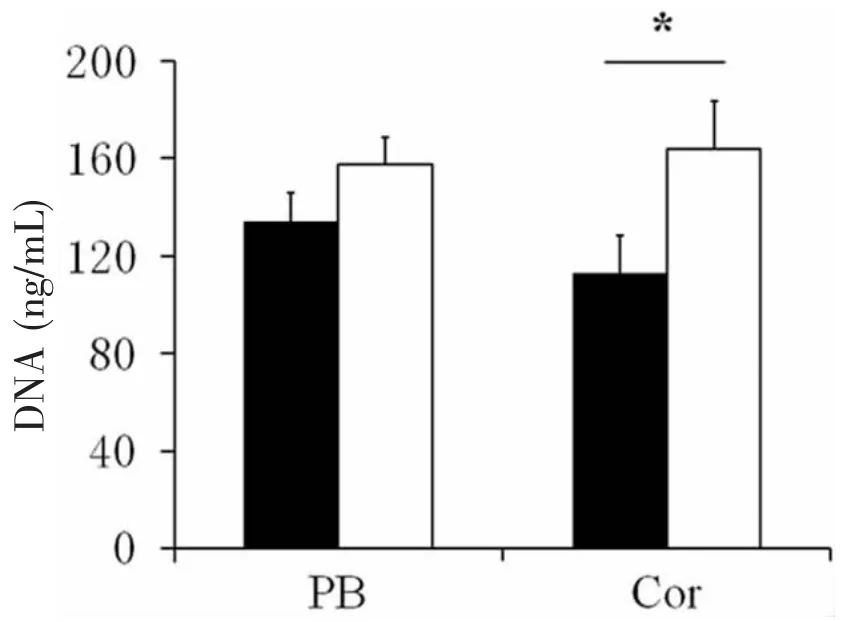
圖5 Fgfr2cC342Y/+小鼠顱縫細胞Picogreen增殖定量分析Fig.5 Picogreen quantification of cell proliferation in Fgfr2cC342Y/+mice
3 討論
FGFR2突變是顱縫早閉癥最常見的基因突變類型(已明確突變類型超過40種),主要見于Crouzon和 Pfeiffer綜合征 Cys342Tyr(C342Y)突變[10]。此外,還見于Pfeiffer綜合征Trp290Cys突變[11]、Beare-Stevenson 綜合征 Tyr375Cys突變[12]、Apert綜合征 S252W[13]、Q289P[14]突變以及 Jackson-Weiss綜合征[15]等。
小鼠模型常用于人顱縫閉合模式研究。本實驗采用Fgfr2cC342Y/+小鼠,結合MicroCT與組織學,作為顱縫閉合模式的常規實驗方法。顱縫閉合分3種模式①開始閉合:MicroCT示顱縫少數位點出現閉合,組織學在不同層面可分別獲取閉合與開放兩種切片;②活躍閉合:MicroCT示顱縫多數位點已經閉合,組織學可獲得閉合與開放兩種切片,以閉合為主;③完全閉合:MicroCT示顱縫縱軸方向及橫斷面(顱骨內層、顱骨外層骨面)均已閉合,組織學結果相同。
本實驗中Fgfr2cC342Y/+小鼠MicroCT與組織學染色結果相符,Fgfr2cC342Y/+雜合子小鼠冠狀縫E18.5開始出現閉合,P0、P1、P5屬于活躍閉合期,P10顱縫完全閉合,而野生型小鼠冠狀縫所有時間點均保持開放,故選擇上述時間點進一步比較閉合與未閉合顱縫間致病基因的表達差異和顱縫細胞增殖礦化情況。
本實驗中,常規選擇 OC(Osteocalcin)、ALP(Alkaline phosphatase)作為標志物。OC由分化晚期的成骨細胞分泌,表達水平逐漸增加,因此可作為成熟成骨細胞的功能標志物[16]。ALP被視為成骨細胞分化的早期標志,隨顱縫閉合逐漸增加[17]。
Rbp4,視黃醇結合蛋白-4,為分泌性血漿蛋白,負責運載肝臟視黃醇向血液、組織傳輸,在組織中催化轉變成視黃酸(RA)[18]。Rbp4在肝外組織中的表達包括脂肪、腎、軟骨、腦組織以及胚胎發育的口腔頜面區域。Rbp4基因敲除小鼠表現出顱骨畸形[19]。Coussens等[16]的前期研究發現,Rbp4定位于人顱縫成骨前緣的顱骨外層骨面骨細胞以及類骨質中,閉合顱縫較未閉合顱縫RBP4表達下調37倍。我們推斷,Rbp4-視黃醇-RA代謝系統的紊亂可引起顱縫早閉癥,可能與FGF/FGFR信號通路相關。
Gpc3,磷脂酰肌醇蛋白聚糖(Gpc)家族成員之一,為膜性硫酸乙酰肝素多糖蛋白,包含核心蛋白、硫酸乙酰肝素鏈(HS)和糖基化磷脂酰肌醇(GPI),Gpc蛋白通過GPI錨定于細胞外膜上[19]。Gpc3在哺乳動物的胚胎期可通過影響多個信號通路而對組織器官的發生和生長發揮重要調控作用,目前主要集中在基因與肝細胞癌相關性的研究[20-21]。前期研究發現,人閉合顱縫Gpc3表達較未閉合顱縫下調7倍。GPC3低表達可能促進顱縫區間充質細胞分化、成骨細胞增殖或抑制細胞凋亡,從而導致顱縫過早閉合。
C1qtnf3,C1Q腫瘤壞死因子相關蛋白-3(又名CTRP3/cartducin,Cors26),在胚胎發育及出生后均可調節軟骨細胞及其前體細胞的增殖[22]。前期研究顯示,C1qtnf3在人顱縫組織中表達,閉合顱縫較未閉合顱縫C1qtnf3表達下調達20倍,通過與軟骨特異性標記物共表達。我們推斷,C1qtnf3除調節顱縫成骨前體細胞的增殖與分化外,尚可能調節軟骨的生成而引起顱縫的閉合。
Fgfr2cC342Y/+小鼠顱縫新致病基因表達分析結果證實,Rbp4、Gpc3、C1qtnf3基因表達模式與小鼠模型與顱縫早閉癥患者Microarray結果相一致,可進一步運用于發病機制的研究。Gpc1、FliI在顱縫閉合中的表達較恒定,野生型與雜合子之間未見明顯統計學差異,提示兩基因可能與顱縫早閉癥無相關性。
本組實驗中MTS和PicoGreen的結果均顯示,Fgfr2cC342Y/+小鼠,Fgfr2功能獲得性突變致使突變型冠狀縫細胞較野生型細胞的增殖量顯著增加,顱縫成骨增強而發生顱縫早閉。此外,本實驗提示MTS試劑對培養細胞具有明顯的毒性。
[1]Virchow R.Untersuchungen uber die entwickelung des schadelgrundes im gesunden und krankhaften zustande,und uber den einfluss derselben auf schadelform,gesichtsbildung und gehirnbau[M].Berlin:G.Reimer,1857.
[2]Coussens AK.Molecular Regulation of calvarial suture morphogenesis and human craniofacial diversity[D].Brisbane:School of Life Sciences,Queensland University of Technology,2007.
[3]Chen L,Li D,Li C,et al.A Ser250Trp substitution in mouse fibroblast growth factor receptor 2(FGFR2)results in craniosynostosis[J].Bone,2003,33(2):169-178.
[4]Wang Y,Xiao R,Yang F,et al.Abnormalities in cartilage and bone development in the Apert syndrome FGFR2(+/S252W)mouse[J].Development,2005,132(15):3537-3548.
[5]Eswarakumar VP,Horowitz MC,Locklin R,et al.A gain-of-function mutation of Fgfr2c demonstrates the roles of this receptor variant in osteogenesis[J].Proc Natl Acad Sci U S A,2004,101(34):12555-12560.
[6]Zhou YX,Xu X,Chen L,et al.A Pro250Arg substitution in mouse Fgfr1 causes increased expression of Cbfa1 and premature fusion of calvarial sutures[J].Hum Mol Genet,2000,9(13):2001-2008.
[7]Livak KJ,Schmittgen TD.Analysis of relative gene expression data using real-time quantitative PCR and the 2[-Delta Delta C(T)]Method[J].Methods,2001,25(4):402-408.
[8]Vandesompele J,De Preter K,Pattyn F,et al.Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes[J].Genome Biol,2002,3(7):RESEARCH0034.
[9]Hellemans J,Mortier G,De Paepe A,et al.qBase relative quantification framework and software for management and automated analysis of real-time quantitative PCR data[J].Genome Biol,2007,8(2):R19.
[10]Reardon W,Winter RM,Rutland P,et al.Mutations in the fibroblast growth factor receptor 2 gene cause Crouzon syndrome[J].Nat Genet,1994,8(1):98-103.
[11]Tartaglia M,Valeri S,Velardi F,et al.Trp290Cys mutation in exon IIIa of the fibroblast growth factor receptor 2(FGFR2)gene is associated with Pfeiffer syndrome[J].Hum Genet,1997,99(5):602-606.
[12]Eun SH,Ha KS,Je BK,et al.The first Korean case of Beare-Stevenson syndrome with a Tyr375Cys mutation in the fibroblast growth factor receptor 2 gene[J].J Korean Med Sci,2007,22(2):352-356.
[13]Girisha KM,Phadke SR,Khan F,et al.S252W mutation in Indian patients of Apert syndrome[J].Indian Pediatr,2006,43(8):733-735.
[14]Freitas EC,Nascimento SR,de Mello MP,et al.Q289P mutation in FGFR2 gene causes Saethre-Chotzen syndrome:some considerations about familial heterogeneity[J].Cleft Palate Craniofac J,2006,43(2):142-147.
[15]Park WJ,Meyers GA,Li X,et al.Novel FGFR2 mutations in Crouzon and Jackson-Weiss syndromes show allelic heterogeneity and phenotypic variability[J].Hum Mol Genet,1995,4(7):1229-1233.
[16]Yousfi M,Lasmoles F,Lomri A,et al.Increased bone formation and decreased osteocalcin expression induced by reduced Twist dosage in Saethre-Chotzen syndrome[J].J Clin Invest,2001,107(9):1153-1161.
[17]Braga V,Gatti D,Rossini M,et al.Bone turnover markers in patients with osteogenesis imperfecta[J].Bone,2004,34(6):1013-1016.
[18]Quadro L,Blaner WS,Salchow DJ,et al.Impaired retinal function and vitamin A availability in mice lacking retinol-binding protein[J].EMBO J,1999,18(17):4633-4644.
[19]Quadro L,Hamberger L,Gottesman ME,et al.Pathways of vitamin A delivery to the embryo:insights from a new tunable model of embryonic vitamin A deficiency[J].Endocrinology,2005,146(10):4479-4490.
Fgfr2cC342Y/+Mouse Model of Crouzon Syndrome in Craniosynostosis
YANG Xianxian1,Jodie T Hatfield2,Susan J Hinze2,MU Xiongzheng3,Peter J Anderson2,Barry C Powell2.1 Department of Plastic and Reconstructive Surgery,Shanghai Ninth People's Hospital,Shanghai Jiaotong University School of Medicine,Shanghai 200011,China;2 Australian Craniofacial Unit,Women's and Children's Hospital,Women's and Children's Health Research Institute,Adelaide,Australia;3 Department of Plastic Surgery,Huashan Hospital,Fudan University School of Medicine,Shanghai 200040,China.
MU Xiongzheng(E-mail:craniomu@gmail.com).
ObjectiveTo investigate a number of novel genes and their molecular mechanisms at play during the premature suture fusion using Fgfr2cC342Y/+mouse model.MethodsThe pattern of cranial suture fusion in Fgfr2cC342Y/+mice(a Cys342Tyr replacement into Fgfr2c to create a gain-of-function mutation equivalent to a mutation in human Crouzon syndromes)were analyzed using microCT and histology.geNorm assay was used to indentify the most stably expressed reference genes in mouse suture tissues and to determine the minimum number of genes required to calculate a reliable normalization factor.Expression of target genes Rbp4,Gpc3,C1qtnf3 etc and markers of osteogenesis were assessed using RT-qPCR during suture fusion and osteogenesis.Comparision of suture cell proliferation in both wildtype and Fgfr2cC342Y/+mice was conducted to analyze the functional basis for the skeletal phenotype of this mutation.ResultsPosterior frontal suture fusion began to occur as early as 10 days in both wildtype and Fgfr2cC342Y/+mice.Fgfr2cC342Y/+mice experienced coronal fusion as early as embryonic 18.5 days and obliterated at 10 days,while wildtype remained patent at all time points.Cyc1,Gapdh and Canx were the most stably expressed housekeeping genes for accurate realtime PCR analysis.A correlation between suture fusion and Rbp4,Gpc3,C1qtnf3 down regulation were demonstrated in Fgfr2cC342Y/+mice.MTS metabolic assay and Picogreen dsDNA quantification analysis showed a significant increase of coronal suture cell proliferation with no change in parietal bone osteoblasts in heterozygote mice.ConclusionSuture fusion in Fgfr2cC342Y/+mice were assessed as a pilot experiment.Fgfr2cC342Y/+mouse model of craniosynostosis syndrome was suitable for molecular mechanism study on suture fusion,particularly for novel gene analysis with FGF signal pathway.Rbp4,Gpc3,C1qtnf3 down regulation during suture fusion was consistent with the human study which suggested may play an important role in bone growth and suture maturation.Fgfr2 gain of function mutation resulted in increased coronal cell proliferation which may lead to increased osteogenesis and premature suture fusion.
Craniosynostosis;Crouzon syndrome;Fgfr2cC342Y/+;RT-qPCR;MicroCT;Rbp4;Gpc3;C1qtnf3
R726.2
A
1673-0364(2012)05-0256-08
10.3969/j.issn.1673-0364.2012.05.004
國家自然科學基金(81171835,81201483);上海市科委非政府間國際合作項目(10410701300)。
200011上海市 上海交通大學醫學院附屬第九人民醫院整復外科(楊嫻嫻);澳大利亞顱頜面外科中心,阿德萊德婦女兒童健康研究中心(Jodie T Hatfield,Susan J Hinze,Peter J Anderson,Barry C Powell);200040 上海市 復旦大學醫學院附屬華山醫院整形外科(穆雄錚)。
穆雄錚(E-mail:craniomu@gmail.com)。
2012年5月14日;
2012年7月30日)
猜你喜歡
興趣閱讀·興趣作文與閱讀(低年級)(2025年8期)2025-08-18 00:00:00
東方少年·布老虎畫刊(2023年8期)2023-08-01 15:45:12
科學大眾(2021年6期)2021-07-20 07:42:44
科學(2020年3期)2020-11-26 08:18:30
學苑創造·A版(2020年9期)2020-10-13 09:41:02
娃娃樂園·綜合智能(2019年3期)2019-04-03 09:17:36
中成藥(2018年2期)2018-05-09 07:19:34
小學生學習指導(低年級)(2017年10期)2017-10-10 01:00:05
湖北師范大學學報(自然科學版)(2015年2期)2016-01-10 08:41:55
云南中醫學院學報(2014年3期)2014-07-31 18:57:34
