三八面體皂石的合成與表征
2012-01-29 02:10:06趙效洪慕旭宏閔恩澤
石油學報(石油加工) 2012年3期
關鍵詞:結構
趙效洪,慕旭宏,閔恩澤
(中國石化石油化工科學研究院石油化工催化材料與反應工程國家重點實驗室,北京100083)
皂石是一種蒙皂石黏土礦物[1],由于具有獨特的結構特征和催化性能而引起人們的廣泛關注[2]。它具有T-O-T三八面體結構[3],因四面體中的硅被鋁取代,酸性比其他天然黏土強[4];層間有可交換離子,經過離子交換、焙燒后形成的層柱皂石的孔徑大于分子篩的十二元環孔,有利于大分子的催化轉化反應;八面體中沒有空位,其熱穩定性高于蒙脫土。因而,皂石是一類很有發展前景的材料。但是,自然界中天然皂石儲量非常低,所含雜質種類較多[5],且其組成因產地不同而有差異,這些都限制了該類材料的應用和發展。合成皂石具有組成均一、性質穩定、酸量可調等優點,是很好的催化材料[6],所以人工合成皂石就顯得格外重要。
合成皂石的研究已有50多年的歷史,其合成的方法都大同小異[7-8]。一般而言,可參照皂石理論結構式按化學計量進行投料,在一定溫度下晶化一定時間,便可得到合成皂石。Kloprogge等[9]指出,水熱溫度是合成高純度皂石的關鍵因素,高溫能為八面體離子提供高的水解速率,有利于皂石的成核和生長。Kuchta等[10]利用SiO2-Al2O3-MgO為原料,在水熱體系中合成了皂石,得到合成皂石的最佳條件為335℃、1.33×107Pa、合成時間7d。Kawi等[7]在285℃下晶化48h成功制得了八面體中含不同Mg/Ni摩爾比的皂石。國內研究人員對皂石合成也開展了一定工作。姜延順[11]采用水熱法于300℃下晶化48h,制得了自然界中不存在的、不同鎵含量的鎵鎳皂石。劉子陽等[12]在285℃下水熱晶化48h,成功制得了層電荷為1.0和2.0的皂石,還對皂石進行了鎵取代研究[13]。盡管合成皂石的研究較多,但還不夠系統全面。筆者采用中高溫水熱合成條件,在堿性環境中合成皂石,并考察了皂石合成的影響因素及其物化性能。
1 實驗部分
1.1 試劑
Na2SiO3·9H2O、NiCl2·6H2O,分析純,國藥集團化學試劑有限公司產品;堿性硅溶膠,質量分數為30%,北京飛龍馬試劑有限公司產品;AlCl3·6H2O,分析純,廣東省汕頭市西隴化工廠產品;MgCl2·6H2O、NaHCO3,分析純,北京益利精細化學品有限公司產品;NaOH,分析純,北京化工廠產品;去離子水,中國石化石油化工科學研究院自制。
1.2 皂石的制備
按理論結構式Nax[Si(8-x)Alx]Ⅳ[Mg6]ⅥO20(OH)4· nH2O的化學計量比投料(x為層電荷),即n(Si)∶n(Al)∶n(Mg)=(8-x)∶x∶6合成鎂皂石[7]。將MgCl2、AlCl3的溶液緩慢倒入Na2SiO3溶液中,用NaOH、NaHCO3調節pH值至13,在一定溫度下,動態攪拌(200r/min)晶化一定時間。經離心分離,固體產物用大量去離子水沖洗,110℃干燥,再用3mol/L NH4Cl于80℃下離子交換2次,110℃干燥,450℃焙燒2h,即得到目的產物鎂皂石。
鎳皂石的理論結構式為Nax[Si(8-x)Alx]Ⅳ[Ni6]ⅥO20(OH)4·nH2O。選用NiCl2作為鎳源,合成條件同上。
1.3 皂石的表征
采用Philips X’pert型X射線衍射儀獲取皂石的XRD譜。采用日本理學電機株氏會社3013型X射線熒光光譜儀進行皂石的元素分析。采用NICOLET6700傅里葉紅外變換光譜儀測定皂石的骨架結構。采用Varian Inova 300M超導核磁共振儀測定皂石的27Al MAS NMR、29Si MAS NMR。采用美國Quantachrome公司AS-6自動吸附儀測定皂石的孔結構和比表面積。采用SDTQ600型差熱分析儀、TA5000DSC2910型差熱分析儀對皂石材料進行TG-DSC表征。采用FEI公司Quanta200型掃描電鏡觀測皂石的形貌(SEM)。采用FEI公司TECNAIGF20(200kV)型透射電鏡獲取皂石的透射電鏡照片(TEM)。
2 結果與討論
2.1 皂石合成的影響因素
2.1.1 晶化溫度的影響
圖1為不同晶化溫度下合成的鎂皂石的XRD譜。從圖1可以看出,晶化溫度為300℃合成的鎂皂石的峰強度最高,(001)晶面的特征衍射峰的對稱性最好,說明晶化溫度越高,鎂皂石的結晶度越高。當晶化溫度高于250℃時,合成的鎂皂石為純相,不存在無定形物質;而晶化溫度為200℃合成的鎂皂石,其(004)晶面的特征衍射峰不明顯,且2θ在20°~30°區域內出現無定形二氧化硅的彌散峰。因此,晶化溫度是合成鎂皂石的一個關鍵因素,高溫有利于合成高結晶度的鎂皂石。
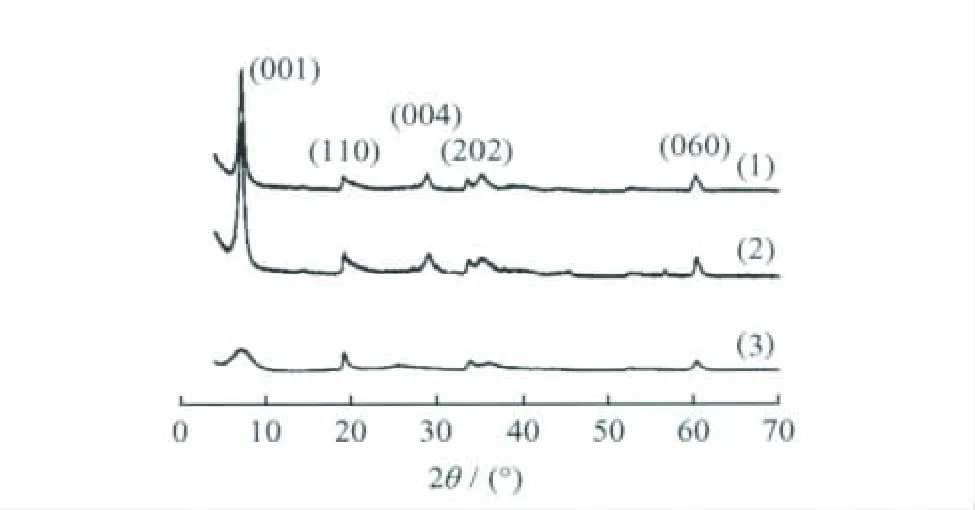
圖1 不同晶化溫度合成的鎂皂石(x=1.5)的XRD譜Fig.1 XRD patterns of Mg-saponites(x=1.5)synthesized at different crystallization temperaturesCrystallization temperature/℃:(1)300;(2)250;(3)200
2.1.2 八面體離子的影響
圖2為采用300℃晶化溫度合成的鎂皂石和鎳皂石的XRD譜。由圖2可以看出,鎳皂石(001)晶面特征衍射峰的強度、對稱性均略高于鎂皂石,說明鎳皂石的有序性略高。但總的來說,二價八面體離子對于皂石合成的影響不大,這一點也與Luca等[14]按配位數計算的結果相吻合。
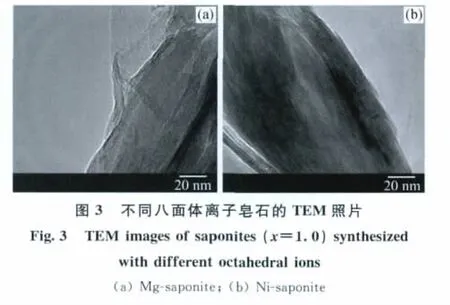
圖3為上述鎂皂石、鎳皂石的TEM照片。由圖3可以看出,鎂皂石的條紋區域較少,且分布不很均勻;鎳皂石的條紋區域較多,且條紋區域排布非常均勻,這也說明鎳皂石的有序性要高于鎂皂石。

2.1.3 n(Si)/n(Al)的影響
圖4為在300℃晶化溫度和不同n(Si)/n(Al)下合成的鎂皂石的XRD譜。從圖4可見,在層電荷從0.38到2.0的范圍內,各鎂皂石都有相似峰形的特征衍射峰,且與標準的鎂皂石峰形一致[4,12]。其中(001)、(060)衍射峰是鎂皂石的特征衍射峰,(060)峰對應的角度為60.5°,說明生成的鎂皂石為三八面體結構[15]。圖4還表明,這些鎂皂石中沒有其他雜質如無定形SiO2。這初步表明,在較寬n(Si)/n(Al)范圍內,均能合成出純相鎂皂石[16-17]。
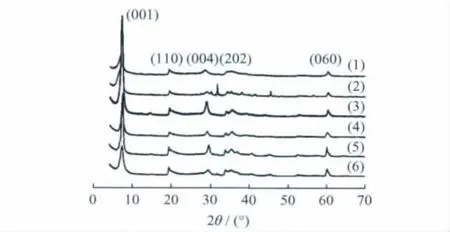
圖4 不同n(Si)/n(Al)合成的鎂皂石的XRD譜Fig.4 XRD patterns of Mg-saponites synthesized with different n(Si)/n(Al)n(Si)/n(Al):(1)7.6/0.4;(2)7.0/1.0;(3)6.8/1.2;(4)6.5/1.5;(5)6.3/1.7;(6)6.0/2.0
2.1.4 晶化時間的影響
圖5為x=1.0時不同晶化時間下合成的鎂皂石的XRD譜。從圖5可以看出,在不同晶化時間下均能合成出鎂皂石;隨晶化時間的延長,合成皂石的結晶度提高。
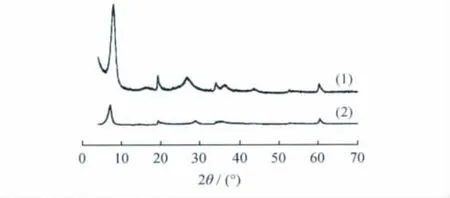
圖5 不同晶化時間合成鎂皂石(x=1.0)的XRD譜Fig.5 XRD patterns of Mg-saponites(x=1.0)synthesized under different crystallization timeCrystallization time/h:(1)48;(2)12
2.2 皂石的物化性質
2.2.1 元素組成
表1為不同層電荷鎂皂石的熒光元素分析的結果。由表1可以看出,盡管合成皂石時按照理論結構式進行投料,但最終按元素分析計算的結構式與理論結構式還是有一定差距。產生這一現象的原因可能來自于熒光測定方法的誤差(±5%)。但總體上,合成皂石的元素組成與理論投料值相近。

表1 不同層電荷(x)鎂皂石的元素分析結果Table 1 Element analysis of Mg-saponites with different layer charge(x)
2.2.2 骨架結構
利用FT-IR可以研究皂石的骨架結構情況,圖6為不同層電荷鎂皂石的FT-IR譜。從圖6可以看出,不同層電荷鎂皂石具有非常相似的FT-IR譜。3447cm-1的吸收峰對應于鎂皂石所包含的水分子的ν(-OH),996/1076cm-1對應于四面體的ν(Si(Al)-O),820cm-1對應于ν(Al-O-Si)或者ν(Al-OH-Mg),656cm-1對應于ν(Mg-O-Al),443cm-1對應于δ(Si-O-Mg)[18]。在1254~1220cm-1和796~784cm-1范圍內沒有吸收峰,這說明鎂皂石產物中沒有無定形SiO2存在[16],這也印證了XRD的結果。
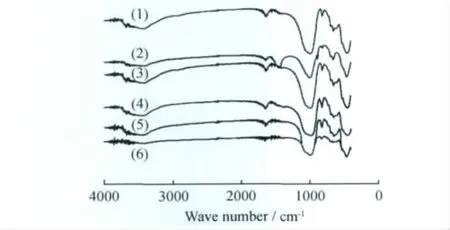
圖6 不同層電荷(x)鎂皂石的FT-IR譜Fig.6 FT-IR spectra of Mg-saponites with different layer charge(x)x:(1)0.4;(2)1.0;(3)1.2;(4)1.5;(5)1.7;(6)2.0
采用對微觀結構敏感的27Al MAS NMR、29Si MAS NMR來考察鎂皂石中Al的配位情況及Si的化學環境,結果示于圖7。從圖7可以看到,不同層電荷的鎂皂石在化學位移64左右均有明顯的共振峰,這剛好對應于四配位鋁。只有層電荷為1.5、1.7的鎂皂石的對稱性不太好,在化學位移為0左右出現較小的共振峰,對應于六配位鋁。這說明合成的皂石原粉中鋁基本是以四配位形式存在的,皂石中鋁的結構很理想。隨著鎂皂石的層電荷的增大,其29Si MAS NMR的特征共振峰的化學位移逐漸增大,對應于Si(nOAl)中n值增大,說明Si原子周圍Al原子的數目增多,這與理論結構也很吻合。
2.2.3 比表面積和孔結構
圖8為不同層電荷鎂皂石樣品的N2吸附-脫附等溫線。由圖8可以看出,此等溫線類型介于Ⅰ型等溫線與Ⅱ型等溫線之間;在p/p0>0.4時,有滯后環存在,說明有介孔存在;從滯后環的形狀來看,鎂皂石中孔道均為狹縫狀孔道,而且隨著層電荷的增加,孔道不對稱性增加[19];在p/p0>0.8時,吸附量迅速增大,說明材料中存在大孔結構,這可能是由鎂皂石層片的邊-面、邊-邊的堆積孔造成的[20]。
表2列出了不同層電荷鎂皂石的比表面積、孔體積和平均孔徑。由表2可以看出,隨層電荷增加,合成鎂皂石的比表面積先增加后降低,孔體積逐漸增加,平均孔徑變化不大。
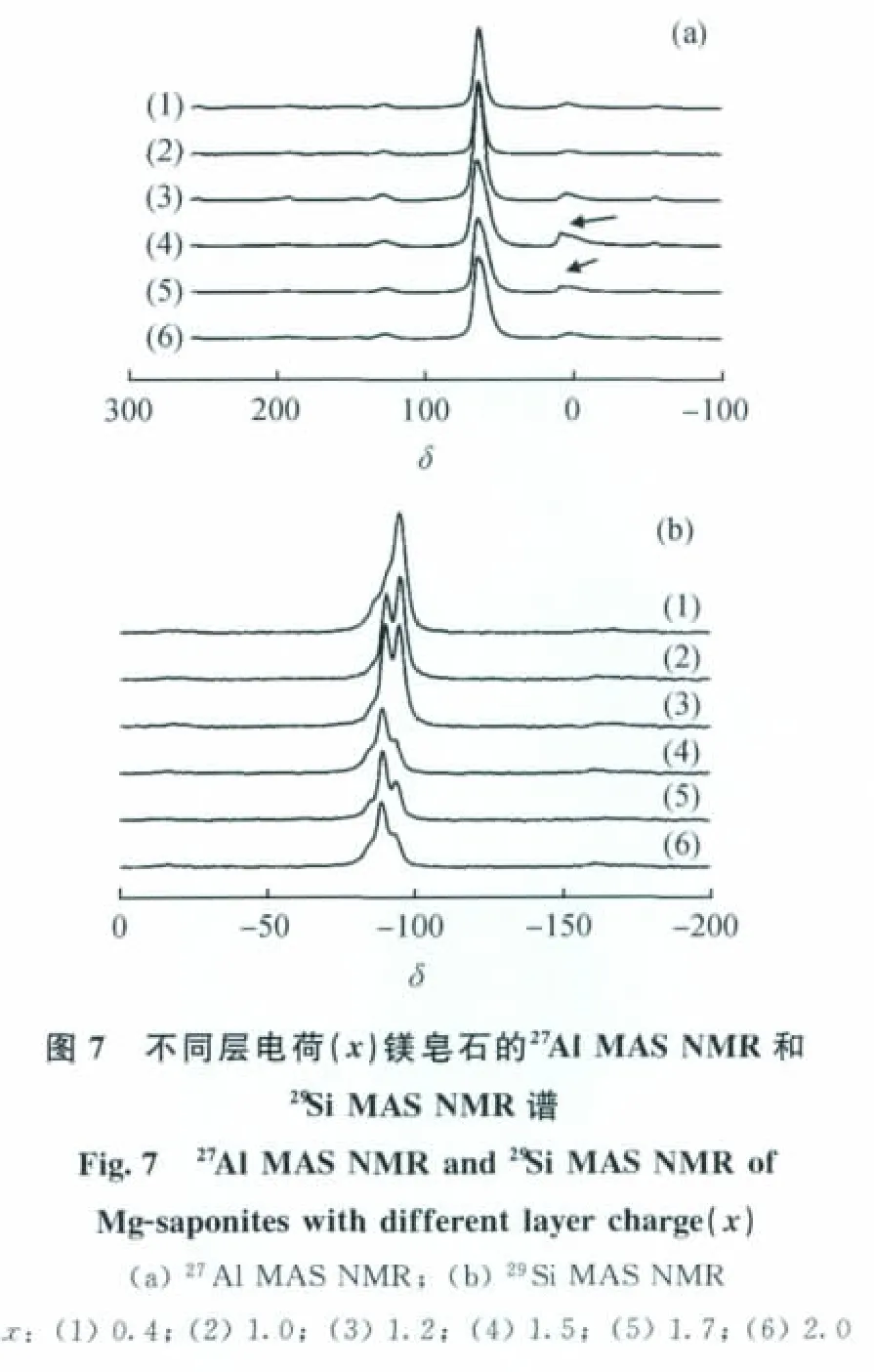
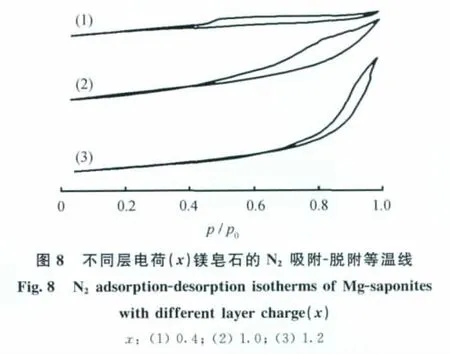

表2 不同層電荷(x)鎂皂石的N2吸附-脫附結果Table 2 Results of N2adsorption-desorption isotherms of Mg-saponites with different layer charge(x)
2.2.4 熱穩定性
以層電荷為1.0的鎂皂石為例,對皂石的熱穩定性進行了研究。圖9為鎂皂石的TG-DSC曲線。由圖9可以看出,DSC曲線共有3次明顯的吸熱、放熱過程,其中175℃吸熱峰對應結晶水的脫除;774℃吸熱峰對應骨架羥基縮合;791℃放熱峰對應層片結構坍塌,這說明鎂皂石的熱穩定性很好,在高于791℃時層片結構才發生坍塌。
2.2.5 形貌
圖10為層電荷為1.0的鎂皂石和鎳皂石的SEM照片。由圖10可以看出,層電荷為1.0的鎂皂石和鎳皂石的外觀形貌均為層片狀,且層片長度不均勻;與鎂皂石相比,鎳皂石的層片更小。
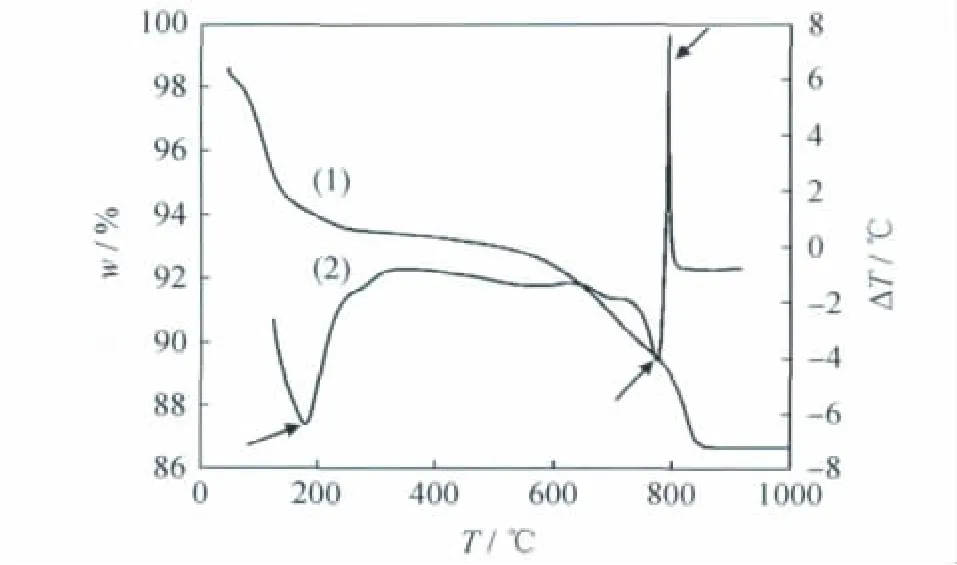
圖9 層電荷為1.0鎂皂石的TG-DSC分析Fig.9 TG-DSC curves of Mg-saponite(x=1.0)(1)TG;(2)DSC

圖10 層電荷為1.0的鎂皂石和鎳皂石的SEM照片Fig.10 SEM images of Mg-,Ni-saponites(x=1.0)(a)Mg-saponite;(b)Ni-saponite
3 結 論
通過人工合成方法成功制備了結構有序性較高的三八面體皂石。晶化溫度是皂石合成最為重要的影響因素,高于250℃可以合成高結晶度的純相皂石;八面體離子對于皂石的有序性有一定影響,鎳皂石的有序性高于鎂皂石;硅/鋁比和晶化時間對皂石的合成影響相對較小。
[1]VOGELS R J M J,KLOPPROGGE J T,GEUS J W.Synthesis and characterization of saponite clays[J].Am Miner,2005,90(5-6):931-944.
[2]DELEVOYE L,ROBERT J L,GRANDJEAN J.23Na 2D3QMAS NMR and29Si,27Al MAS NMR investigation of Laponite and synthetic saponites of variable interlayer charge[J].Clay Miner,2003,38:63-69.
[3]DING Z,KLOPROGGE J T,FROST R L,et al.Porous clays and pillared clays-based catalysts 2Areview of the catalytic and molecular sieve applications[J].J Porous Mater,2001,8(4):273-293.
[4]YAO M,LIU Z Y,WANG K X,et al.Synthesis and characterization of pillared high layer charged synthetic saponite[J].J Porous Mater,2004,11:229-238.
[5]UTRACKI L A,SEPEHR M,BOCCALERI E.Review:Synthetic,layered nanoparticles for polymeric nanocomposites(PNCs)[J].Polym Adv Technol,2007,18(1):1-37.
[6]TRUJILLANO R,VICENTE M A,RIVES V,et al.Preparation,alumina-pillaring and oxidation catalytic performances of synthetic Ni-saponite[J].Microp Mesop Mater,2009,117(1-2):309-316.
[7]KAWI S,YAO Y Z.Saponite catalysts with systematically varied Mg/Ni ratio:Synthesis,characterization,and catalysis[J].Microp Mesop Mater,1999,33(1-3):49-59.
[8]GRANQUIST W T,COUNTY A.Synthetic silicate minerals:US,3252757[P].1962-07-27.
[9]KLOPROGGE J T,KOMARNENI S,AMONETTE J E.Synthesis of smectite clay minerals:A critical review[J].Clays Clay Miner,1999,47(5):529-554.
[10]KUCHTA L,FAJNOR V S.Optimal conditions for hydrothermal synthesis of saponite[J].Chem Papers,1988,42(3):339-345.
[11]姜廷順 .鎵鎳皂石的合成、交聯、表征及催化性能的研究[J].江蘇理工大學學報(自然科學版),1999,20(6):57-60.(JIANG Tingshun.Studies on the synthesis,cross linking,characterization and catalytic properties of GaNi substituted saponite[J].Journal of Jiangsu University of Science and Technology(Natural Science),1999,20(6):57-60.
[12]劉子陽,蔣大振,張愛君 .皂石的合成、表征及其催化性能[J].高等學校化學學報,1991,12(4):502-505.(LIU Ziyang,JIANG Dazhen,ZHANG Aijun,et al.Synthesis,characterization and catalytic activity of saponites and their pillared analogues[J].Chem J Chinese Universities,1991,12(4):502-505.)
[13]劉子陽,黃東律,孫鐵,等 .鎵取代皂石的合成與交聯研究[J].高等學校化學學報,1992,13(4):517-520.(LIU Ziyang,HUANG Donglü,SUN Tie,et al.Studies on the synthesis and cross-linking of Gasubstituted saponite[J].Chem J Chinese Universities,1992,13(4):517-520.)
[14]LUCA V,MACLACHLAN D J,HOWE R F,et al.Synthesis and characterization of a(Zn,Ti)-substituted layered silicate[J].J Mater Chem,1995,5(4):557-564.
[15]POLVEREJAN M,PAULY T R,PINNAVAIA T J.Acidic porous clay heterostructures(PCH):Intragallery assembly of mesoporous silica in synthetic saponite clays[J].Chem Mater,2000,12(9):2698-2704.
[16]RODRIGUEZ M A V,GONZALEZ J D L,MUNOZ M A B,et al.Preparation of microporous solids by acid treatment of a saponite[J].Microp Mater,1995,4:251-264.
[17]AVILA L R,FARIA E H,CIUFFI K J,et al.New synthesis strategies for effective functionalization of kaolinite and saponite with silylating agents[J].Colloid Inter Sci,2010,341:186-193.
[18]PRIHOD’KO R,SYCHEV M,HENSEN E J M,et al.Physicochemical and catalytic characterization of nonhydrothermally synthesized Mg-,Ni-and Mg Ni-saponitlike material[J].Microp Mesop Mater,2004,69:49-63.
[19]徐如人,龐文琴,于吉紅,等 .分子篩與多孔材料化學[M].北京:科學出版社,2005:148.
[20]XIA Y D,MOKAYA R.Crystalline-like molecularly ordered mesoporous aluminosilicates derived from aluminosilica-surfactant mesophases via benign template removal[J].J Phys Chem B,2006,110(18):9122-9131.
猜你喜歡
小獼猴智力畫刊(2023年4期)2023-04-23 08:49:58
哲學評論(2021年2期)2021-08-22 01:53:34
中華詩詞(2019年7期)2019-11-25 01:43:04
模具制造(2019年3期)2019-06-06 02:10:54
中學生數理化·高一版(2018年1期)2018-02-10 05:20:03
影視與戲劇評論(2016年0期)2016-11-23 05:26:01
七彩語文·寫字與書法(2016年7期)2016-07-28 21:40:22
七彩語文·寫字與書法(2016年6期)2016-07-15 19:36:34
人間(2015年21期)2015-03-11 15:23:21
現代企業(2015年9期)2015-02-28 18:56:50
