濁點萃取-原子光譜法在金屬離子形態分析中的應用
2012-08-06 00:32:02楊利寧趙玲玲陳建榮
浙江師范大學學報(自然科學版) 2012年4期
關鍵詞:檢測
楊利寧,趙玲玲,陳建榮,2
(1.浙江師范大學化學與生命科學學院,浙江金華 321004;2.浙江師范大學地理與環境科學學院,金華浙江 321004)
隨著生命科學、生物工程和環境科學等學科的迅速發展,分析對象日益復雜多樣,對復雜基體中痕量和超痕量組分的分離和檢測成為突出的問題.雖然現代儀器分析方法的檢出限越來越低,但要直接分析某些組分的含量,也常會遇到困難,有時甚至是不可能的.這是因為:一方面是樣品本身的物理化學狀態有的不適合直接測定,或者分析方法對極低含量的組分靈敏度不夠;另一方面是存在基體干擾,或者缺乏相應的校正標準和試劑.因此,必須借助各種各樣的分離富集技術,以提高分析方法的靈敏度和選擇性.濁點萃取作為一種分離富集的手段,具有萃取效率高、富集因子大、操作簡便、安全、經濟和便于實現聯用等優點.一般來說,任何與膠束體系直接反應(一般指疏水有機物)或預先衍生(例如金屬離子與合適的螯合劑的螯合反應)后的物質,都可以用濁點萃取法從其溶液中萃取和富集.濁點萃取法(CPE)與原子光譜法結合進行分離富集和檢測痕量金屬離子是一種有效解決上述問題的方法.本文著重介紹了近十年來濁點萃取-原子光譜法在測定不同形態金屬離子含量方面的應用.
1 濁點萃取
濁點萃取技術是利用表面活性劑在水溶液中特殊的相行為對溶質進行分離的.表面活性劑分子在水溶液中能夠形成親水基團向外、憎水基團向內的膠團.表面活性劑膠團溶液可以使微溶于水或不溶于水的有機物的溶解度大大增加,稱作表面活性劑溶液的增溶.同時,某些表面活性劑水溶液在加熱到一定溫度(濁點溫度)以上后變渾濁并出現分相.濁點萃取法就是利用表面活性劑溶液的增溶(solubilization)和分相(phase separation)來實現溶質的富集和分離.濁點萃取法使用的各種類型的表面活性劑中,開發最早、應用最廣泛的是非離子表面活性劑.具有代表性的非離子表面活性劑是聚氧乙烯烷基醚(polyoxy-ethylene alkyl ether,POEAE),它是在濁點萃取中最常應用的非離子表面活性劑.非離子表面活性劑溶液在加熱到濁點溫度以上時出現分相,分為表面活性劑富集的凝聚相和表面活性劑含量很低的主體水相.表1列出了一些聚氧乙烯烷基醚類表面活性劑的濁點溫度.可以看出,濁點溫度與表面活性劑分子的烷基鏈長和聚氧乙烯鏈長有密切的關系.當烷基鏈長相同時,濁點溫度隨聚氧乙烯鏈長的增加而升高;相反,聚氧乙烯鏈長相同時,濁點溫度隨烷基鏈長的增加而降低.
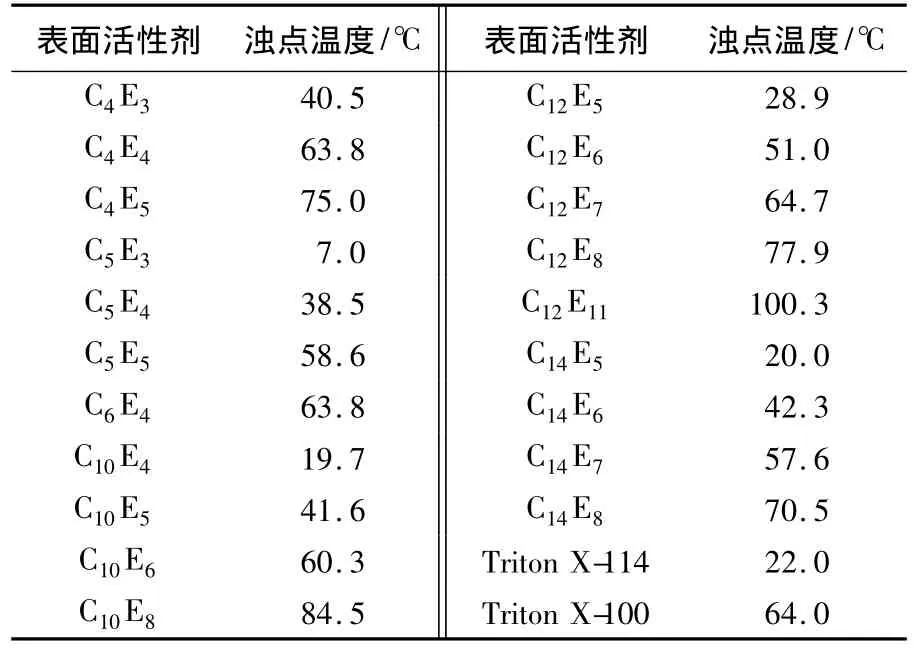
表1 聚氧乙烯烷基醚類表面活性劑的濁點溫度[1]
文獻[2]對于直脂肪鏈聚氧乙烯烷基醚類的表面活性劑進行了研究,給出了濁點溫度(T,℃)與表面活性劑結構之間的經驗關系
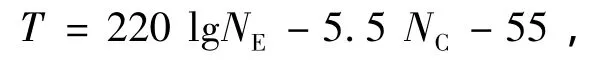
其中,NE和NC分別是表面活性劑分子中的聚氧乙烯鏈和脂肪鏈中的碳原子個數.
非離子表面活性劑溶液的溫度升高時,表面活性劑中聚氧乙烯基的氧原子與水形成氫鍵的能力減弱,表面活性劑在水中的溶解度降低,進而使表面活性劑的臨界膠團濃度變小,膠團的聚集數(aggregate number)增大,膠團體積增大.當溶液溫度到達濁點溫度時,溶液由于膠團體積過大而混濁分相.
濁點分相可以解釋為聚氧乙烯鏈的結構變化導致氧乙烯單元的極性降低、親水性下降,在濁點溫度以上膠團與水的混合熵的大量減少.表面活性劑溶液體系的Gibbs自由能由2個部分組成:膠團在溶液中均勻分布的熵(entropie effect)增加造成的自由能降低和溶液中膠團之間的相互作用(internal energy effect)造成的自由能增加.這2種作用的競爭造成了非離子表面活性劑溶液特有的濁點分相現象.
值得注意的是,在水相中,表面活性劑的濃度并不為零,而是等于或者大于表面活性劑的臨界膠團濃度.因此,在主體水相中,表面活性劑分子形成的各種結構的膠團仍然存在,并且這些膠團對于溶質仍然具有增溶作用.溫度變化導致的表面活性劑溶液的分層現象是一個可逆的過程,降溫之后體系會恢復成澄清的一相.兩相分層需要借助兩相之間的密度差完成,一般需要對體系進行離心分離或者重力沉降.
2 濁點萃取法與原子光譜法聯用
濁點萃取法作為痕量元素分離富集的手段,在分析化學中廣泛應用,它能與多種原子光譜檢測技術聯用,包括原子吸收光譜法、等離子體原子發射光譜法、原子熒光光譜法等.
2.1 濁點萃取-原子吸收光譜法
火焰原子吸收光譜法(FAAS)的進樣速度快,測定時間短,在0.5 min內即可測定一個樣品,并且標準曲線的線性可以達到0.999,重現性也比較好,相對誤差小于1%.但火焰原子吸收光譜法的進樣體積相對來說較大,并且濁點萃取之后的表面活性劑相是粘滯的,因此需要加入含一定量硝酸的甲醇溶液降低其黏度,這就降低了濁點萃取的富集倍數.
與火焰原子吸收光譜法相比,石墨爐原子吸收光譜法(GFAAS)更受到青睞.因為石墨爐原子吸收光譜法的精確度高、靈敏度高,檢測限可達到10-13g·mL-1,比火焰原子吸收光譜法的檢出限更低,并且石墨爐原子吸收光譜法可以采用自動進樣技術,操作簡便快捷,只需一人操作即可完成全部測定過程,進樣量也只有幾μL,這在提高濁點萃取的富集倍數方面有很大的優勢.由于石墨爐進樣量小,所以經過濁點萃取后的表面活性劑相不需要用有機溶劑稀釋,可直接用移液槍注入石墨管內部進行測定,也可采用自動進樣裝置注入,表面活性劑還會改善溶液和石墨管的接觸性能,提高原子化速度.將CPE與電熱原子吸收光譜法(ETAAS)聯用,靈敏度比CPE-FAAS法提高2~3個數量級.
2.2 濁點萃取-等離子體原子發射光譜法
等離子體原子發射光譜法(ICP-AES)雖然是一種常用的痕量金屬的檢測方法,但其檢出限仍然高于樣品中金屬的濃度,為了獲得準確、可靠的結果,需對樣品中的被測物進行富集和分離,以便直接檢測.原子發射光譜法(AES)有如下特點:由于各元素同時發射各自的特征光譜,所以AES可多元素同時檢測,并且選擇性高;AES的進樣速度快,分析速度快;檢出限較低;準確度較高,尤其是定性分析,目前在各種分析手段中是最有效的.
由此,前處理技術——濁點萃取法與低檢出限的原子發射光譜法聯用,形成了一種對金屬的富集和檢測的有效技術,適用于測定環境樣品中的痕量金屬元素.
2.3 濁點萃取-原子熒光光譜法
有些元素的原子蒸氣在輻射能激發下會產生熒光,原子熒光光譜法(AFS)就是通過測量待測元素發射的熒光強度對待測元素進行定量分析的,根據熒光譜線的波長還可以進行定性分析.原子熒光光譜法的儀器設備簡單、檢測靈敏度高、熒光光譜干擾少、線性范圍寬、可以進行多元素測定.該方法也是測定各種環境樣品的標準檢驗推薦方法,在農業、醫學、地球化學、地質、材料和環境科學等領域的應用非常廣泛.
3 濁點萃取-原子光譜法在金屬離子形態分析中的應用
根據國際純粹與應用化學聯合會(IUPAC)的定義[3],形態分析是指測定樣品中某元素的不同鍵合形式、不同氧化態和該元素的總量.不同形態離子的作用和毒性大不相同.例如:鉻的主要存在形態是Cr(Ⅲ)和Cr(Ⅵ),Cr(Ⅲ)是人體中必不可少的微量元素,而 Cr(Ⅵ)被認為是毒性物質[4],其毒性比Cr(Ⅲ)大100倍;無機錫主要以Sn(Ⅱ)和Sn(Ⅳ)的形式存在于環境樣品中,由于無機錫是一種耐腐蝕的金屬,因而在食品包裝和電子工業中具有重要應用.然而,不同形態的元素總量不能為其毒性、生物利用和轉化、循環方式等提供充足的信息.因此,需要對元素的不同形態進行分析測定.
濁點萃取法是近年來金屬離子形態分析中常用的方法,在實際應用中,例如與光譜法聯用時,有2種方法:一是采用差減法;二是調節溶液pH值或加入不同螯合劑的方法.
3.1 測定 Cr
鉻元素廣泛存在于地殼中,其在各種工業工藝品和產品中的廣泛應用是環境中鉻污染的主要來源.元素鉻的生物學和毒理學特性很大程度上取決于其化學形態,其中:Cr(Ⅲ)參與糖代謝,是維持動物正常的葡萄糖耐量、生長及壽命不可缺少的;但Cr(Ⅵ)對人體有毒害性[4],主要是偶然吸入極限量的鉻酸或鉻酸鹽后,引起腎臟、肝臟、神經系統和血液的廣泛病變,導致死亡.濁點萃取分離富集鉻形態時多采用氧化還原法(即差減法),也有采用不同螯合劑的方法.有關濁點萃取與原子光譜法聯用分離富集測定 Cr(Ⅲ)和Cr(Ⅵ)的研究進展[5-20]見表2.
3.2 測定 Sb
文獻[21]采用N-苯甲酰苯基羥胺和TritonX-114體系萃取 Sb(Ⅲ),用 L-半胱氨酸還原Sb(Ⅴ)以達到萃取測定總Sb的目的,濃縮相經甲醇稀釋后用FAAS測定,差減法計算Sb(Ⅴ)的量.文獻[22]利用 pH 5.0 時 APDC 對 Sb(Ⅲ)的選擇性萃取達到與Sb(Ⅴ)分離,同樣采用L-半胱氨酸還原Sb(Ⅴ)測定銻的總量.用ETAAS測定,Sb(Ⅲ)的檢出限達 0.02 μg·L-1,方法成功應用于食品接觸材料浸出液中銻形態的測定.文獻[23]利用APDC與Sb(Ⅲ)的螯合反應濁點萃取Sb(Ⅲ),分相后不經離心分離,萃取混合液經棉花填充的小柱吸附表面活性劑濃縮相后,再以100 μL乙腈洗脫,洗脫液用電熱蒸發等離子體原子發射光譜(ETV-ICP-AES)測定;然后以L-半胱氨酸還原 Sb(Ⅴ)測定總銻,再以差減法計算Sb(Ⅴ)的量.該方法的靈敏度更高、檢測限更低,并成功用于水樣中無機銻形態的分析測定.
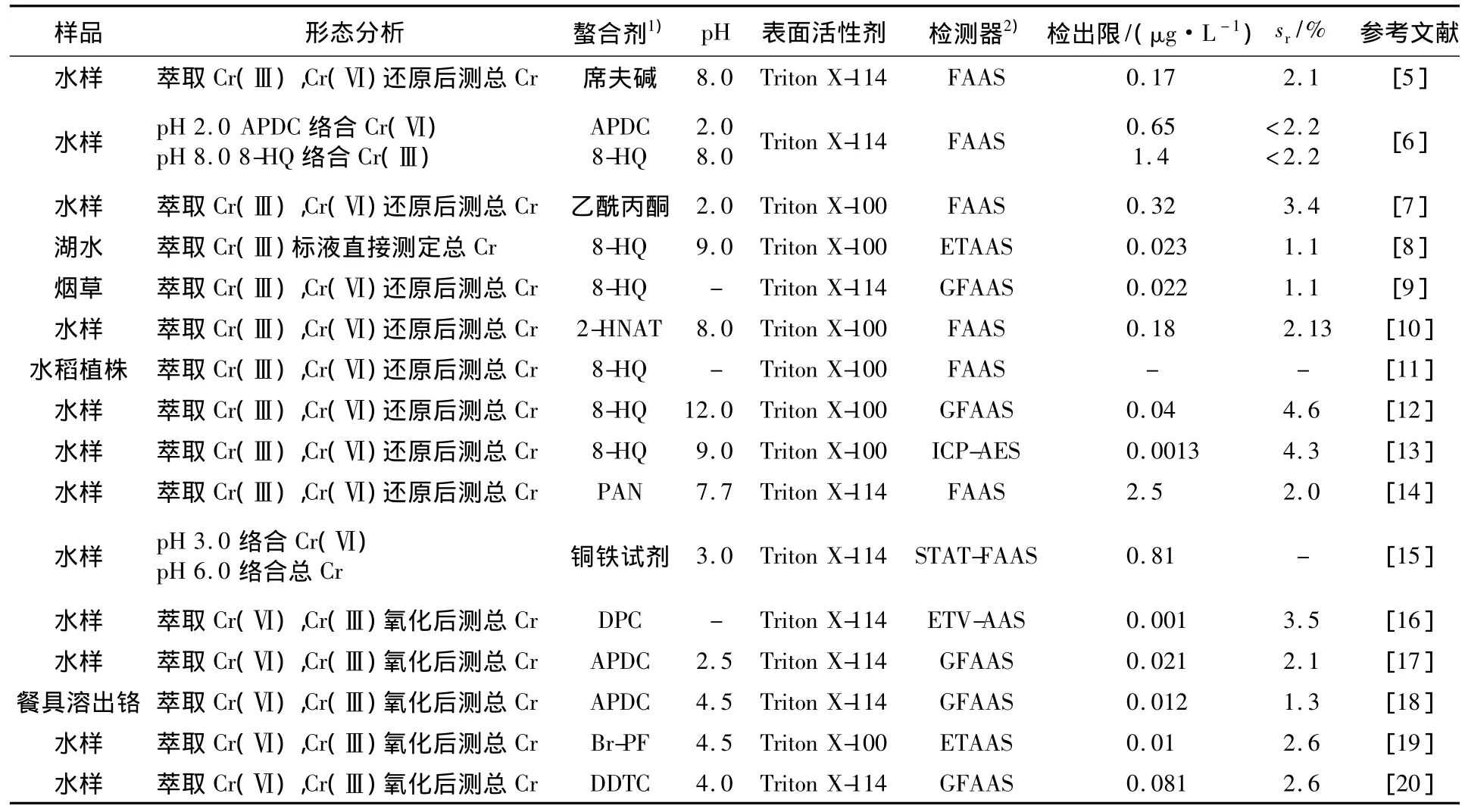
表2 濁點萃取與原子光譜法聯用測定Cr(Ⅲ)和Cr(Ⅵ)
3.3 測定 Se
Güler等[24]利用在酸性條件下 2,3-二氨基萘(DAN)對 Se(Ⅳ)的選擇性螯合萃取達到與Se(Ⅵ)分離的目的,用12 mol·L-1HCl還原Se(Ⅵ)測定Se的總量,用熒光光譜法測定.方法用于測定硒酸鹽、亞硒酸鹽及總無機硒含量均達到 μg·L-1級.Sounderajan 等[25]利用 3,3'-二氨基聯苯胺(DAB)和 Triton X-114體系萃取Se(Ⅳ),富集因子可達100,用4 mol·L-1HCl微波加熱還原Se(Ⅵ)以測定總Se,用ETAAS測定,環境水樣中 Se 的檢測限達 0.002 5 μg·L-1,并且可用于一些動物食物中總Se的測定.
3.4 測定 As
張力培等[26]利用 pH 3.0 時 As(Ⅲ)與 APDC絡合形成疏水性螯合物被萃取到表面活性劑相,用鎢絲電熱原子熒光光譜法測定,從而實現與As(Ⅴ)的分離測定;利用Na2S2O3還原As(Ⅴ)測定總As,然后由差減法求得As(Ⅴ)的含量.As(Ⅲ)的檢出限為 0.9 μg·L-1,可用于水樣中As(Ⅲ)和As(Ⅴ)的測定.文獻[27]利用鉬酸鹽與As(Ⅴ)的絡合反應,用濁點萃取-ETAAS測定As(Ⅴ),KMnO4氧化 As(Ⅲ)以測定總 As,然后差減法求得As(Ⅲ)的含量,方法的檢測限達到0.01 μg·L-1,富集倍數達 52.5.該方法成功地用于自來水樣中As(Ⅴ)和As(Ⅲ)的測定及生物樣品中總As的測定.
3.5 測定 Fe
Giokas等[28]將流動注射分析-光度法和FAAS結合開展鐵形態的研究,以APDC同時螯合濁點萃取Fe(Ⅲ)和Fe(Ⅱ),并以FAAS測定2種形態鐵的總量,然后以流動注射分析-光度法測定Fe(Ⅱ),總量減去Fe(Ⅱ)的量即為Fe(Ⅲ)的值.該方法避免了其他類似分步形態分析方法需對某一形態進行氧化或還原的步驟,同時,因為采用了流動注射分析技術,方法相對簡便快速.該方法成功應用于環境水樣中鐵形態的測定.Shakerian等[29]采用如上的方法,利用鐵試劑ferron同時與Fe(Ⅲ)和Fe(Ⅱ)在高于濁點溫度時生成的螯合物萃取到表面活性劑相,用流動注射-FAAS測定了總鐵.
3.6 測定 Sn
Gholivanda等[30]利用 α-多金屬氧酸鹽在 pH 1.2時僅與 Sn(Ⅱ)螯合,而在 pH 3.6 時與Sn(Ⅱ)和Sn(Ⅳ)同時反應的特點,通過控制酸度分別對Sn總量和Sn(Ⅱ)萃取富集,用FAAS法測定,Sn(Ⅱ)和Sn(Ⅳ)的檢出限分別為8.4和12.6 μg·L-1,富集因子可達100.文獻[31]在非離子表面活性劑存在下,將Sn(Ⅳ)與鈣試劑羧酸鈉(CCA)反應萃取,Sn(Ⅱ)在酸性條件下經高錳酸鉀氧化為Sn(Ⅳ)以測定總Sn量,差減法計算Sn(Ⅱ)的量,火焰原子吸收光譜法測定,Sn(Ⅳ)富集因子達 100,檢出限 2.86 μg·L-1.
3.7 測定其他元素
文獻[32]基于I-存在下Hg(Ⅱ)與甲基綠生成不溶于水的離子締合物,被萃取到表面活性劑相中,從而與水相中的甲基汞MeHg+分離,包含Hg(Ⅱ)的膠束相引入電感耦合等離子體發射光譜儀(ICP-OES)檢測,從而實現了 Hg(Ⅱ)和MeHg+的直接形態分析.肖宇等[33]建立了不需形成螯合物的濁點萃取-石墨爐原子吸收光譜法測定水中Tl(Ⅲ)的新方法,方法的檢測限為0.018 μg·L-1,加標回收率為 98.0% ~101%,方法適用于水中痕量Tl(Ⅲ)的測定.陳海婷等[34]采用APDC為絡合劑,建立了濁點萃取預富集電熱原子吸收光譜法測定水中痕量鉈的方法,方法的富集倍率達到31,可用于自來水和河水中痕量鉈的測定.
4 展望
濁點萃取技術與原子光譜法聯用在金屬離子形態分析方面已經得到廣泛的應用,濁點萃取法比傳統的液-液萃取法有很多優點,例如:避免了大量使用有毒的有機溶劑;擁有更高的回收率和富集因子等.但濁點萃取在水浴、離心、分離表面活性劑相時還是要花費一定的時間,目前還沒有達到快速檢測、現場檢測的要求.近幾年,在線流動注射濁點萃取的方法有所發展,因為流動注射省去了所有的人工操作,但在聯用技術方面有所限制,還沒有大范圍地推廣.如果把濁點萃取流動注射與原子光譜法的聯用技術進一步發展、優化,這樣既可以節省樣品預處理的時間,又可以達到快速檢測的目的,在金屬離子形態分析方面有很好的應用前景.另外,利用色譜柱的分離功能雖然可以同時測定不同金屬形態的含量,但是色譜法一般檢出限較高,達不到痕量檢測的目的.因此,這兩方面的研究還需要進一步深入.
[1]莫小剛,劉尚營.非離子表面活性劑濁點的研究進展[J].化學通報,2001(8):483-487.
[2]Gu Tiren,Sj?blom J.Surfactant structure and its relation to the Krafft point,cloud point and micellization:some empirical relationships[J].Colloids Surf,1992,64(1):39-46.
[3]Templeton D M,Ariese F,Cornelis R,et al.Guidelines for terms related to chemical speciation and fractionation elements.Definitions,structural aspects,and methodological approaches(IUPAC Recommendations 2000)[J].Pure Appl Chem,2000,72(8):1453-1470.
[4]陳杭亭,曹淑秦,曾憲津.電感耦合等離子體質譜方法在生物樣品分析中的應用[J].分析化學,2001,29(5):592-600.
[5]Shemirani F,Abkenar S D,Mirroshandel A A,et al.Preconcentration and speciation of chromium in water samples by atomic absorption spectrometry after cloud-point extraction[J].Anal Sci,2003,19(10):1453-1456.
[6]Paleologos E K,Stalikas C D,Tzouwara-Karayanni S M,et al.Micelle-mediated methodology for speciation of chromium by flame atomic absorption spectrometry[J].J Anal At Spectrom,2000,15(3):287-291.
[7]Sun Zhimei,Liang Pei.Determination of Cr(Ⅲ)and total chromium in water samples by cloud point extraction and flame atomic absorption spectrometry[J].Microchimi Acta,2008,162(1/2):121-125.
[8]朱霞石,江祖成,胡斌,等.濁點萃取-電熱原子吸收光譜法分析鉻的形態[J].分析化學,2003,31(11):1312-1316.
[9]Zhu Xiashi,Hu Bin,Jiang Zucheng.Cloud point extraction combined with graphite furnace atomic absorption spectrometry for the determination of chromium species and their distribution in cigarette and cigarette ash[J].Int J Environ Anal Chem,2004,84(12):927-934.
[10]Kiran K,Kumar K S,Prasad B,et al.Speciation determination of chromium(Ⅲ)and(Ⅵ)using preconcentration cloud point extraction with flame atomic absorption spectrometry(FAAS)[J].J Hazard Mater,2008,150(3):582-586.
[11]李銘芳,盧志紅,黃喜根.原子吸收光譜法測定水稻植株中不同部位鉻的形態[J].江西農業大學學報,2006,28(5):797-799.
[12]肖宇,張克榮,鄭波.濁點萃取-石墨爐原子吸收法測定水中痕量鉻和銀[J].中國衛生檢驗雜志,2006,16(6):667-668.
[13]李靜,梁沛,施踏青.濁點萃取預富集ICP-AES測定水樣中痕量鉻[J].廣西師范大學學報:自然科學版,2003,21(1):59-60.
[14]Matos G D,dos Reisa E B,Costa A C S,et al.Speciation of chromium in river water samples contaminated with leather effluents by flame atomic absorption spectrometry after separation/preconcentration by cloud point extraction[J].Microchem J,2009,92(2):135-139.
[15]盧菊生,田久英,吳宏.濁點萃取-石英雙縫管捕集火焰原子吸收光譜法分析鉻價態[J].分析化學,2009,37(1):99-102.
[16]Ezoddin M,Shemirani F,Khani R.Application of mixed-micelle cloud point extraction for speciation analysis of chromium in water samples by electrothermal atomic absorption spectrometry[J].Desalination,2010,262(1/2/3):183-187.
[17]方炳華.濁點萃取-石墨爐原子吸收光譜法測定環境水樣中痕量鉻(Ⅵ)和總鉻[J].浙江師范大學學報:自然科學版,2008,31(4):437-440.
[18]周賽春,江海亮,周先波,等.濁點萃取-石墨爐原子吸收光譜法對不銹鋼餐具溶出鉻的形態分析[J].分析測試學報,2008,27(4):405-407.
[19]Zhu Xiashi,Hu Bin,Jiang Zucheng,et al.Cloud point extraction for speciation of chromium in water samples by electrothermal atomic absorption spectrometry[J].Water Res,2005,39(4):589-595.
[20]祝旭初,沈慧明,沃燕娜.濁點萃取-GFAAS測定水中鉻的形態[J].光譜實驗室,2006,23(4):762-765.
[21]Fan Zhefeng.Speciation analysis of antimony(Ⅲ)and antimony(Ⅴ)by flame atomic absorption spectrometry after separation/preconcentration with cloud point extraction[J].Microchim Acta,2005,152(1/2):29-33.
[22]Jiang Xiuming,Wen Shengping,Xiang Guoqiang.Cloud point extraction combined with electrothermal atomic absorption spectrometry for the speciation of antimony(Ⅲ)and antimony(Ⅴ)in food packaging materials[J].J Hazard Mater,2010,175(1/2/3):146-150.
[23]Li Yingjie,Hu Bin,Jiang Zucheng.On-line cloud point extraction combined with electrothermal vaporization inductively coupled plasma atomic emission spectrometry for the speciation of inorganic antimony in environmental and biological samples[J].Anal Chim Acta,2006,576(2):207-214.
[24]Güler N,Maden M,Bakirdere S,et al.Speciation of selenium in vitamin tablets using spectrofluorometry following cloud point extraction[J].Food Chem,2011,129(4):1793-1799.
[25]Sounderajan S,Kumar G K,Udas A C.Cloud point extraction and electrothermal atomic absorption spectrometry of Se(Ⅳ)-3,3'-diaminobenzidine for the estimation of trace amounts of Se(Ⅳ)and Se(Ⅵ)in environmental water samples and total selenium in animal blood and fish tissue samples[J].J Hazard Mater,2010,175(1/2/3):666-672.
[26]張力培,吳鵬,何靚,等.濁點萃取鎢絲電熱蒸發原子熒光光譜分析法測定水樣中的As(Ⅲ)和As(Ⅴ)[J].四川大學學報:自然科學版,2007,44(3):646-648.
[27]Shemirani F,Baghdadi M,Ramezani M.Preconcentration and determination of ultra trace amounts of arsenic(Ⅲ)and arsenic(Ⅴ)in tap water and total arsenic in biological samples by cloud point extraction and electrothermal atomic absorption spectrometry[J].Talanta,2005,65(4):882-887.
[28]Giokas D L,Paleologos E K,Karayannis M I.Speciation of Fe(Ⅱ)and Fe(Ⅲ)by the modified ferrozine method,FIA-spectrophotometry,and flame AAS after cloud-point extraction[J].Anal Bioanal Chem,2002,373(4/5):237-243.
[29]Shakerian F,Dadfarnia S,Shabani A M H.Separation,Preconcentration and measurement of inorganic iron species by cloud point extraction and flow injection flame atomic absorption spectrometry[J].J Iran Chem Soc,2009,6(3):594-601.
[30]Gholivanda M B,Babakhaniana A,Rafiee E.Determination of Sn(Ⅱ)and Sn(Ⅳ)after mixed micelle-mediated cloud point extraction using α-polyoxometalate as a complexing agent by flame atomic absorption spectrometry[J].Talanta,2008,76(3):503-508.
[31]Songül Ulusoy,Halil íbrahim Ulusoy,Mehmet Ak?ay,et al.Inexpensive and versatile method for trace Sn(Ⅱ)and Sn(Ⅳ)ions in food samples by CPE/FAAS[J].Food Chem,2012,134(1):419-426.
[32]Li Yingjie,Hu Bin.Sequential cloud point extraction for the speciation of mercury in seafood by inductively coupled plasma optical emission spectrometry[J].Spectrochimica Acta Part B:Atomic Spectroscopy,2007,62(10):1153-1160.
[33]肖宇,鄭波,張克榮.非螯合物濁點萃取-石墨爐原子吸收法測定水中痕量鉈(Ⅲ)[J].分析化學,2007,35(11):1654-1656.
[34]陳海婷,陳建國,林力,等.濁點萃取電熱原子吸收光譜法測定水中痕量鉈[J].分析試驗室,2009,28(11):60-62.
猜你喜歡
中國設備工程(2022年12期)2022-07-11 04:33:00
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:36
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:34
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:50
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:48
