高效液相色譜法測定硫酸長春新堿的有關物質
2012-09-17 06:40:50龍亞秋羅超華
中國藥業 2012年3期
李 華 ,龍亞秋 ,羅超華 ,楊 芳
(1.南方醫科大學南方醫院,廣東 廣州 510515; 2.廣州中醫藥大學第二附屬醫院,廣東 廣州 510120;3.南方醫科大學中醫藥學院,廣東 廣州 510515)
硫酸長春新堿為抗腫瘤類原料藥,中國藥典中其有關物質測定采用高效液相色譜法[1]。由于產品工藝中的起始原料發生變更,可能帶入新雜質,為此筆者參閱歐洲藥典(EP 7.0版)和美國藥典(USP 34版),對中國藥典所述有關物質的測定方法進行了驗證。現報道如下。
1 儀器與試藥
島津LC-20A型高效液相色譜儀,LC Solution色譜工作站;Sartorius-BP210型電子天平。硫酸長春新堿對照品、硫酸長春堿對照品(中國藥品生物制品檢定所,批號為100130-200502);硫酸長春新堿原料藥(廣州綠葉化工有限公司,批號為100701,100702,100703);甲醇為色譜純,水為純化水,其余均為分析純。
2 方法和結果
2.1 色譜條件
色譜柱:Shiseido C8柱(250 mm ×4.6 mm,5 μm);流動相:取二乙胺15 mL,加水985 mL,用磷酸調pH至7.5,作為流動相A,甲醇為流動相B,進行梯度洗脫(0~12 min時A體積分數為38%,12~27 min時A體積分數由38%線性變化至8%,27~29 min時,A體積分數由8%線性變化至38%,29~34 min時維持A體積分數為38%);檢測波長:297 nm;柱溫:30℃;流速:1.5 mL/min。
2.2 溶液制備
取本品,精密稱定,加水溶解并稀釋制成每1 mL中約含1 mg的溶液,作為高濃度供試品溶液;精密量取1 mL,置25 mL量瓶中,用水稀釋至刻度,搖勻,作為低濃度供試品溶液。取硫酸長春新堿及硫酸長春堿對照品適量,用水溶解并稀釋制成每1 mL分別含有1 mg的混合溶液,作為系統適用性溶液。
2.3 方法學考察
系統適用性試驗:在擬訂的色譜條件下,精密吸取系統適用性溶液20 μL,注入液相色譜儀。結果硫酸長春新堿的保留時間約為15 min,硫酸長春堿峰與硫酸長春新堿峰的分離度應大于4.0,見圖 1。
專屬性試驗:取水(溶劑空白)20 μL 進樣,記錄色譜圖,結果表明溶劑對雜質測定無干擾。取系統適用性溶液和高濃度供試品溶液各20 μL進樣,記錄色譜圖,結果硫酸長春堿峰與硫酸長春新堿主峰之間的分離度大于4.0,硫酸長春新堿峰與相鄰各雜質峰之間的分離度均大于1.5(見表 1 和表 2、圖 1),表明方法的專屬性較好。
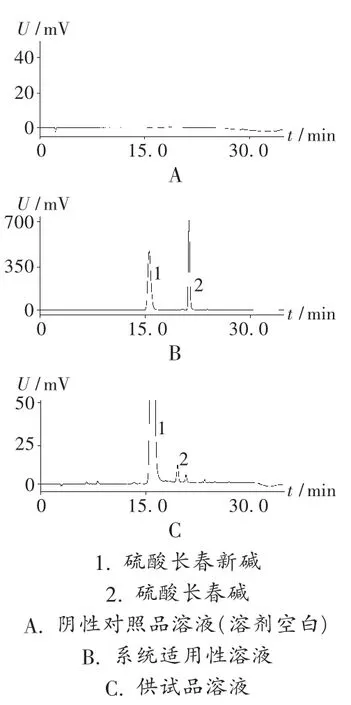
圖1 高效液相色譜圖

表1 已知物質的分離度測試
線性關系考察:取硫酸長春堿對照品約25 mg,精密稱定,用水溶解并稀釋至質量濃度為0.1 g/L,作為標準貯備液A;精密吸取標準貯備液A 1 mL,用水稀釋至10 mL,質量濃度0.01 g/L,作為標準貯備液B。取硫酸長春新堿樣品約0.1 g,精密稱定,置10 mL容量瓶中,用水稀釋至刻度,混合均勻,質量濃度約為10 g/L,作為標準貯備液 C。分別精密吸取標準貯備液 B 0.5,1.0,5.0 mL 和標準貯備液 A 1,2,4,5 mL,各置 10 mL 容量瓶中,精密吸取1 mL,用水稀釋至刻度,混合均勻,得到編號1-7的標準溶液。取1.0 mL標準貯備液C,置10 mL量瓶中,加水稀釋至刻度,搖勻,作為高濃度樣品溶液;取1.0 mL高濃度樣品溶液,置25 mL量瓶中,加水稀釋至刻度,搖勻,作為低濃度樣品溶液。取編號1-7的標準溶液,分別進樣,每個質量濃度連續進樣3次。以硫酸長春堿為代表雜質,其實際質量濃度在 0.510 ~51.04 μg/mL,(相當于0.05% ~5.0%)范圍內與峰面積呈良好的線性關系,線性回歸方程為 Y=0.000 01 X -0.147 5,r=0.999 98。

表2 樣品存在雜質的分離度測試
精密度試驗:分別取硫酸長春堿、硫酸長春新堿對照品溶液,重復進樣6次。結果峰面積的 RSD硫酸長春堿為0.20%(n=6),硫酸長春新堿為 0.11%(n=6),均小于 2.0% ,表明方法精密度高。
重復性試驗:取同一份樣品共6份,依法測定。結果峰面積的RSD硫酸長春堿為0.24%(n=6),硫酸長春新堿為0.14%(n=6),均小于2.0%,表明方法重復性良好。
加樣回收試驗:稱取已知含量的的原料藥6份,分別精密加入對照品適量,依法制備溶液并測定含量,計算回收率。結果見表3。硫酸長春新堿的回收率均在98.0% ~102.0%范圍內,符合要求。
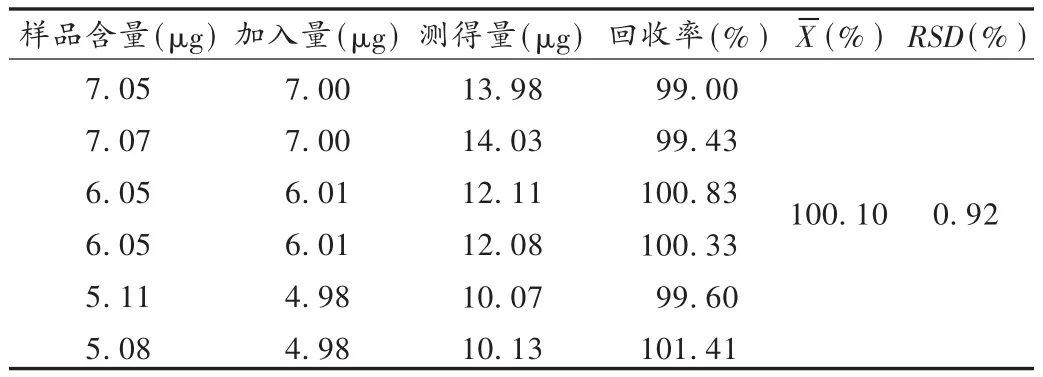
表3 硫酸長春新堿加樣回收試驗結果(n=6)
檢測限及定量限確定:該方法的定量限和檢測限以硫酸長春堿作為雜質進行評估。取線性關系考察項下編號1和2的標準溶液逐步稀釋,分別進樣,記錄色譜圖。當待測組分的信噪比為2~3倍,對應該組分的最小檢測質量濃度為0.51 μg/mL,相當于0.05%;當信噪比10~20倍時,對應該組分的最小定量質量濃度為 1.02 μg/mL,相當于 0.1%。結果表明,該方法的靈敏度足以用于硫酸長春新堿雜質的檢測。
穩定性試驗:取同一樣品,依法制備供試品溶液,于2,4,8,12,24 h時分別進樣。結果硫酸長春堿與硫酸長春新堿峰面積響應值的 RSD分別為0.35%和0.17%(n=5),表明該方法所用溶液穩定性良好。
可變參數耐用性試驗:取系統適用性溶液(含有硫酸長春新堿及其主要雜質硫酸長春堿),除可變的條件外,其他參數均與常規分析條件一致,依法進樣,記錄色譜圖。結果見表4。可見,對流動相比例、柱溫、流速、流動相pH作一定程度的改變時,均可維持硫酸長春堿與硫酸長春新堿分離度大于4的系統適用性要求,表明方法的耐用性較好。
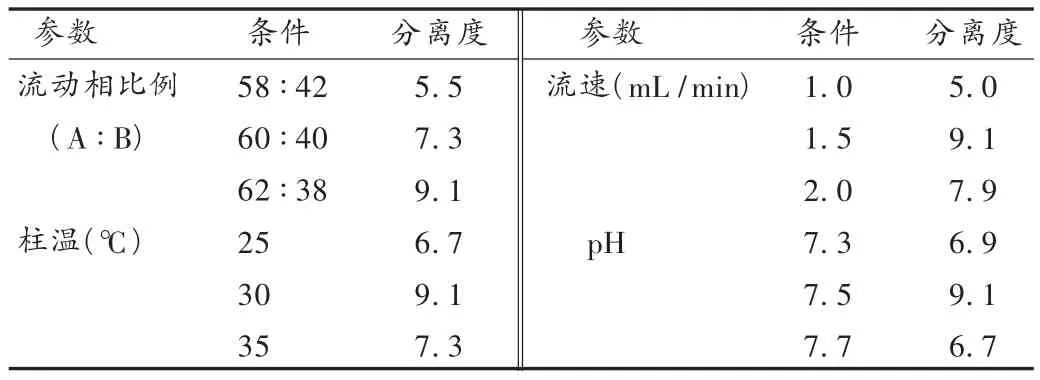
表4 耐用性試驗結果
2.4 樣品有關物質測定
取起始原料變更前后的3批樣品,按2.2項下方法制備供試品溶液,在擬訂的色譜條件下進行分析,記錄色譜圖,按自身對照法以峰面積計算雜質含量。結果見表5。可見,起始原料變更后無新雜質產生,原有雜質含量比起始原料變更前雜質含量有所降低。
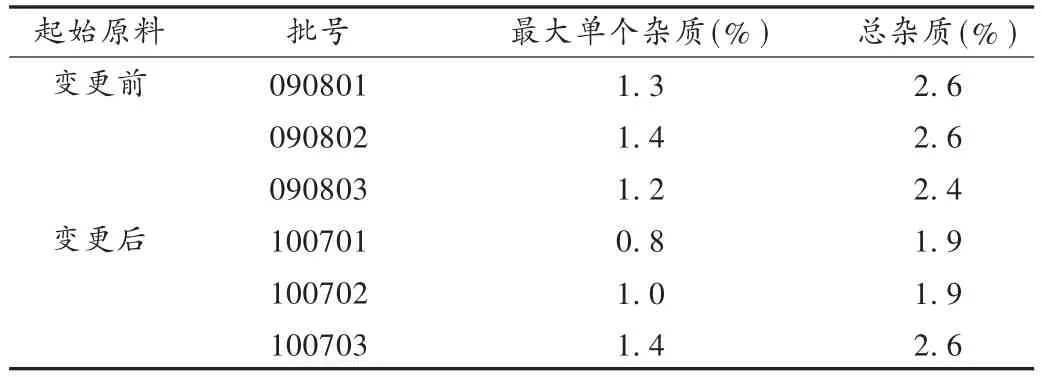
表5 硫酸長春新堿的有關物質含量測定結果
3 討論
本試驗針對硫酸長春新堿原料藥的起始物料來源發生變更,按現行版藥典的要求,對中國藥典收載的硫酸長春新堿有關物質檢測方法進行了詳盡而全面的方法學考察,重點考察專屬性與耐用性。結果表明,該方法專屬性、重復性良好,方法準確,檢測靈敏度高,耐用性好,可用于起始原料變更后的產品有關物質檢測。
在專屬性考察中,除關注已知雜質硫酸長春堿與主分硫酸長春新堿的分離情況,更特別關注樣品中實際存在的雜質與主成分的分離度,試驗結果符合測定要求。此外,還從溶液穩定性、流動相比例、溫度、流速和pH等幾個方面考察了方法的耐用性,測定結果令人滿意。通過方法學考察,為該產品雜質質量控制方法的可靠性和適用性提供了充分的實驗依據。
[1]國家藥典委員會.中華人民共和國藥典(二部)[M].北京:中國醫藥科技出版社,2010:953-954.
