R-(+)-α-硫辛酸的合成工藝
2012-10-19 02:24:10冷一欣牛錦森黃春香
化工進展 2012年6期
關鍵詞:催化劑
冷一欣,牛錦森,黃春香
(常州大學石油化工學院,江蘇 常州 213164)
研究開發
R-(+)-α-硫辛酸的合成工藝
冷一欣,牛錦森,黃春香
(常州大學石油化工學院,江蘇 常州 213164)
以6,8-二氯辛酸為原料,S-(-)-α-苯乙胺為拆分劑進行拆分反應得到R-(+)-6,8-二氯辛酸;經酯化制得R-(+)-6,8-二氯辛酸乙酯;后經硫化水解一步合成R-(+)-α-硫辛酸。考察了投料比、溶劑、催化劑、溫度等條件對產物收率、比旋光度的影響。結果表明:拆分反應較佳條件為n[S-(-)-α-苯乙胺]∶n(6,8-二氯辛酸)=0.45∶1,溶劑為乙酸乙酯;酯化反應較佳條件為催化劑為對甲苯磺酸,反應時間7 h;硫化反應較佳條件為溫度為65 ℃;相轉移催化劑用量0.4 g,總收率為44.3%。通過紅外光譜、比旋光度、核磁共振等對產物和中間產物進行了表征。
6,8-二氯辛酸;拆分;酯化;硫化;R-(+)-α-硫辛酸
α-硫辛酸(α-lipoic acid),化學名為1,2-雙硫環戊烷基-3-戊酸,有一個手性中心,兩種對映異構體中R-(+)-α-硫辛酸的生理活性遠高于S-(-)-α-硫辛酸。α-硫辛酸能夠消除致病的自由基[1],屬于維生素類藥物[2],是唯一兼具脂溶性與水溶性的萬能抗氧劑[3-4]。對于肝病、糖尿病、艾滋病、皮膚癌、帕金森氏征、風濕病等多種疾病有治療功效[5-6]。
1983年,Golding等[7]成功確定了R-(+)-α-硫辛酸的構型。Elliott等[8]首次在手性輔助試劑的誘導下,進行了不對稱合成[9],成功合成出R-(+)-α-硫辛酸,總收率37%,該反應所用的原料及試劑極其昂貴,反應條件苛刻,工業上難以操作。Gopalan等[10]首次利用微生物酶催化合成R-(+)-α-硫辛酸,總收率10%,但微生物法[11]中酶的純度不高并具有弱致病性,至今未見有用于大規模工業生產的報道。
國內研究者普遍采用化學拆分法[12],但直接拆分外消旋α-硫辛酸法[13]得到的產品中含有殘留的有毒拆分劑且總收率較低。本研究在此基礎上改進,采用拆分外消旋6,8-二氯辛酸得到R-(+)-6,8-二氯辛酸,經酯化、硫化、水解合成R-(+)-α-硫辛酸。拆分后的S-(-)-6,8-二氯辛酸較穩定,可以消旋化再利用,此法反應條件溫和、收率高、成本低,具有工業應用前景。
1 實驗部分
1.1 試劑與儀器
6,8-二氯辛酸(江蘇神州工業有限公司,純度≥96%);S-(-)-α-苯乙胺(工業級,金壇華陽化工廠);二硫化鈉(硫化鈉法自制,含量≥95%);對甲苯磺酸(AR,國藥集團化學試劑公司);四正丁基溴化銨(AR,國藥集團化學試劑公司);無水乙醇(AR,國藥集團化學試劑公司,含量≥99.7%),其余試劑均為國藥,AR。
Avance 500 MHz核磁共振波譜儀 (德國Bruker公司);Waters-717高效液相色譜儀(美國Waters公司);PROTéGé460型傅里葉紅外光譜儀(美國Nicolet公司);WZZ-2B型數字式自動旋光儀(上海浦東物理光學儀器廠);X-4型顯微熔點測定儀(北京第三光學儀器廠);RE-2000型旋轉蒸發儀(上海亞榮生化儀器廠)。
1.2 合成路線
本研究的合成路線如圖1所示。
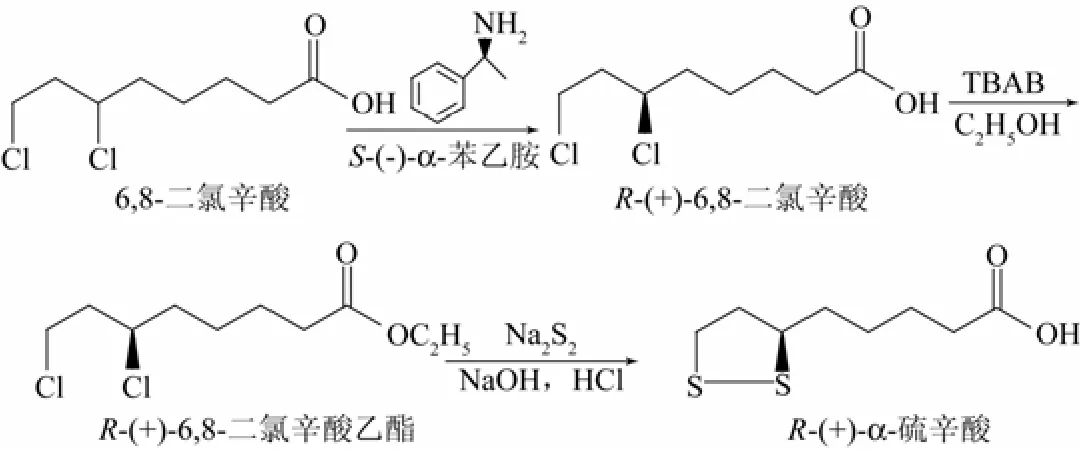
圖1 R-(+)-α-硫辛酸的合成路線
1.3 實驗方法
1.3.1 R-(+)-6,8-二氯辛酸的合成
將20.0 g(0.094 mol)6,8-二氯辛酸加入三口燒瓶中,20 mL乙酸乙酯作為溶劑,在溫度35 ℃,攪拌條件下滴加S-(-)-α-苯乙胺5.5 g(0.045 mol),反應6 h。反應液冷卻靜置,得到乳白色的晶塊,抽濾(母液保留),乙酸乙酯洗滌,真空干燥,得到白色的絮狀固體。將其溶于水,經鹽酸(5 mol/mL)酸化,乙酸乙酯萃取,干燥,脫溶,得到淡黃色的油狀液體7.5 g。純度(HPLC)97.0%,收率72.8%。比旋光度:[α]20D=26.4(c=1,EtOH),[文獻[14]值[α]20D=26.7(c=1,EtOH)]。IR(cm-1):2947.9(νO-—H),2867.5(νC—H),1708.4(νC=O),665.1(νC—Cl)。1H NMR(CDCl3,500 MHz,δ),1.42~1.46(m,2H,4位CH2),1.50~1.62(m,2H,5位CH2),1.66~1.83(m,2H,3位CH2),2.01~2.16(m,2H,7位CH2),2.24~2.30(m,2H,2位CH2),3.66~3.74(m,2H,8位CH2),4.10~4.15(m,1H,6位CH)。
1.3.2 R-(+)-6,8-二氯辛酸乙酯的合成
將5.0 g(0.023 mol)R-(+)-6,8-二氯辛酸加入三口燒瓶中,加入80 mL無水乙醇和0.5 g對甲苯磺酸,充分攪拌,回流溫度下反應8 h;乙酸乙酯萃取,20 mL水洗滌3次,干燥,脫溶得到淡黃色的油狀液體5.5 g。純度(HPLC)96.4%,收率93.7%。比旋光度:[α]20D=25.5(c=1,EtOH),[文獻[14]值[α]20D=26.5(c=1,EtOH)]。IR(cm-1):2942.9(νO—H),2867.3(νC—H),1734.3(νC=O),665.1(νC—Cl)。1H NMR(CDCl3,500 MHz,δ),1.16~1.19(t,J=7.5 Hz,3H,乙基中的CH3),1.42~1.48(m,2H,4位CH2),1.51~1.63(m,2H,5位CH2),1.72~1.84(m,2H,3位CH2),2.05~2.18(m,2H,7位CH2),2.27(t,J=6.9Hz,2H,2位CH2),3.70~3.76(m,2H,8位CH2),4.02~4.05(q,J=7.4Hz,2H,乙基中的CH2),4.13~4.15(m,1H,6位CH)。
1.3.3 R-(+)-α-硫辛酸的合成
將5.0 g(0.021 mol)R-(+)-6,8-二氯辛酸乙酯加入三口燒瓶中,加入50 mL無水乙醇,0.3 g正四丁基溴化銨(TBAB),在溫度65 ℃下,按n[R-(+)-6,8-二氯辛酸乙酯∶n(Na2S2)=1∶1加入Na2S2溶液,攪拌反應6 h;降溫至55 ℃,滴加10.0 g NaOH(20%)溶液,水解反應2 h后停止。HCl(5 mol/mL)酸化,甲苯萃取、干燥、脫溶,得到黃色的油狀液體,環己烷溶解重結晶,經抽濾得到淡黃色的粉末晶體3.7 g。熔點:46~51 ℃(文獻[15]值46~48 ℃),純度(HPLC)97.3%,收率71.4%。比旋光度:[α]20D=117.2(c=1,EtOH),[文獻[16]值[α]20D=119.1(c=1,EtOH)]。IR(cm-1):2927.5(νO—H),2865.1(νC—H),1692.1(νC=O),1249.6和1201.9(νC—S),673.1(νS—S)。1H NMR(CDCl3,500 MHz,δ):3.60~3.55(m,1H,6位CH),3.21~3.10(m,2H,8位CH2),2.50~2.44(m,1H,7位CH2),2.39~2.37(t,2H,2位CH2),1.95~1.88(m,1H,7位CH2),1.75~1.60(m,2H,3位CH2),1.60~1.54(m,2H, 5位CH2),1.54~1.42(m,2H,4位CH2)。
1.3.4 產品的純度分析條件
色譜條件:固定相Ultimate TM HPLC Column(XB-C18,5 μm,4.6mm×150 mm),柱箱溫度25℃,UV檢測波長330 nm,進樣量20 μL;流動相為甲醇-水-乙酸(300∶200∶0.5,體積比),流速1.0 mL/min。
2 結果與討論
2.1 R-(+)-6,8-二氯辛酸的合成
2.1.1 投料比對拆分反應的影響
20 mL乙酸乙酯作溶劑,S-(-)-α-苯乙胺緩慢滴加,溫度35 ℃下,攪拌反應6 h,測定產物R-(+)-6,8-二氯辛酸的收率和比旋光度,得到其隨S-(-)-α-苯乙胺與6,8-二氯辛酸摩爾比變化的曲線,結果見圖2。
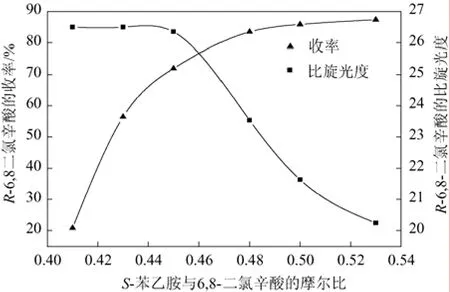
圖2 投料比對拆分反應的影響
外消旋體6,8-二氯辛酸存在對映異構體,通過與S-(-)-α-苯乙胺作用形成非對映異構體[R-(+)-6,8-二氯辛酸·S-(-)-α-苯乙胺],酸處理得到R-(+)-6,8-二氯辛酸,投料比對其收率和比旋光度都有較大的影響。由圖2可以看出,隨著S-(-)-α-苯乙胺與6,8-二氯辛酸摩爾比的增加,反應收率呈線性增長,最后達到穩定;過量的S-(-)-α-苯乙胺會結合S-(-)-6,8-二氯辛酸形成另一種非對映異構體,產品的比旋光度則呈遞減曲線,在0.45∶1后陡降,說明S-(-)-α-苯乙胺結合了相反構型的對映異構體,所以投料比以0.45∶1為宜。
2.1.2 溶劑對反應的影響
溫度35 ℃下,以n[S-(-)-α-苯乙胺]:n(6,8-二氯辛酸)= 0.45∶1投料,加入20 mL溶劑,攪拌反應6 h,分別考察不同溶劑對R-(+)-6,8-二氯辛酸收率及比旋光度的影響,結果見表1。
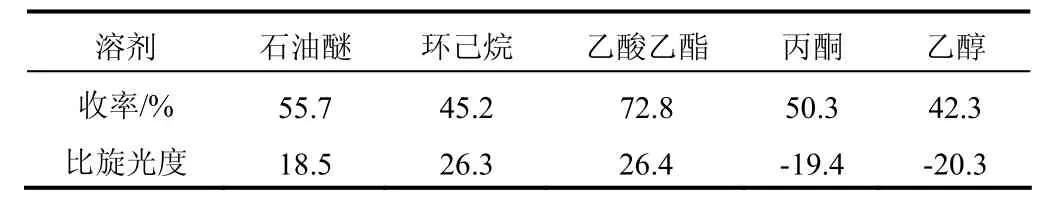
表1 溶劑對拆分反應的影響
手性拆分劑對相應構型的優先選擇,與非對映異構體晶鹽中的氫鍵網絡結構變化有關系,而氫鍵網絡是由溶劑的介電性決定的。溶劑分子通過填充非對映異構體鹽的空間使其析出。表1中,丙酮和乙醇作溶劑時產物的比旋光度是負數,可能由于丙酮、乙醇的介電常數較大導致拆分試劑選擇了S構型的對映體;石油醚和環己烷作溶劑時產物的收率不高,原因是溶劑化效果不好,晶鹽不能很好析出;乙酸乙酯作溶劑,既可以對消旋混合物中的一種對映異構體進行手性識別[17];又可以通過溶劑化[18]作用使優先構型的微溶非對映異構體結晶析出。因此,乙酸乙酯作溶劑為宜。
2.2 R-(+)-6,8-二氯辛酸乙酯的合成
2.2.1 催化劑對反應的影響
投入5.0 g R-(+)-6,8-二氯辛酸和80 mL無水乙醇,回流溫度下反應8 h,分別加入1.0 g不同種類的催化劑,考察其對R-(+)-6,8-二氯辛酸乙酯收率及比旋光度的影響,結果見表2。
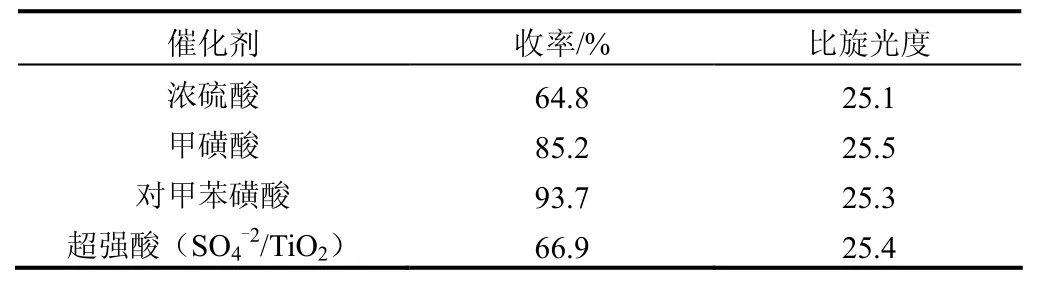
表2 催化劑種類對酯化反應的影響
在酯化反應中酸作為催化劑的作用是將羰基質子化,正電性較強的羰基更有利于親核試劑醇的進攻。由表2可知,濃硫酸和甲磺酸為催化劑時收率較低,原因是其酸性較強,致使底物酸發生脫水、脫羥基等副反應。超強酸(SO4-2/TiO2)之所以能起催化作用,源于SO4-2在Ti表面配位吸附使Ti—O的電子云偏移,路易斯酸中心得到強化,同時Ti的空d軌道與醇中氧原子上的孤電子對發生配位作用產生質子酸中心。酯化反應中超強酸(SO4-2/TiO2)與C2H5OH中的氧原子配位,發生副反應,生成內酯使得收率降低。對甲苯磺酸由于酸性適中,不容易發生副反應,收率較高。催化劑種類對產物的比旋光度基本沒有影響,因此,對甲苯磺酸作為酯化反應的催化劑較好。
2.2.2 反應時間對酯化反應的影響
投入5.0 g R-(+)-6,8-二氯辛酸和80 mL無水乙醇,加入0.5 g對甲苯磺酸作為催化劑,考察不同的反應時間對R-(+)-6,8-二氯辛酸乙酯收率及比旋光度的影響,結果見圖3。
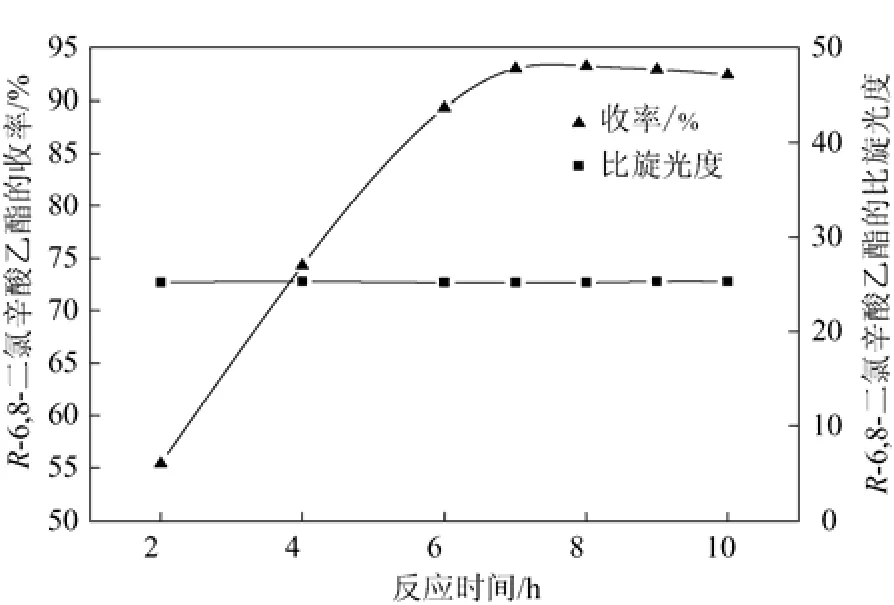
圖3 反應時間酯化反應的影響
由圖3可知,在反應7h時間內,隨著反應時間的延長,R-(+)-6,8-二氯辛酸乙酯的收率不斷提高,當反應時間為7 h時,收率達到最大93.2%。7 h以后反應收率趨于平衡且略有下降,可能是發生了一些副反應所致。整個時間段R-(+)-6,8-二氯辛酸乙酯的比旋光度呈平穩趨勢,基本不受時間的影響。因此,反應時間確定為7 h為宜。
2.3 R-(+)-α-硫辛酸的合成
2.3.1 溫度對硫化反應的影響
以50 mL無水乙醇為溶劑,0.3 g四丁基溴化銨(TBAB)為相轉移催化劑,在投料比n[R-(+)-6,8-二氯辛酸乙酯]∶n(Na2S2)=1∶1,硫化反應6 h、55℃水解反應2 h條件下,考察溫度對R-(+)-α-硫辛酸的收率及比旋光度的影響,結果見表3。
此反應屬于親核取代,—S—S—鍵先進攻位阻較小的8位碳原子,另一端隨后進攻6位碳原子,形成了活化能較低的五元環,溫度為其鍵斷裂提供了必要的能量。由表3可知,在55~65 ℃內,R-(+)-α-硫辛酸的收率隨著溫度的升高而增加,比旋光度穩定;當反應溫度達到65 ℃時,R-(+)-α-硫辛酸的收率最高達70.1%;反應溫度超過65 ℃時,收率降低同時比旋光度減小,這是因為在較高溫度下,雙硫鍵聚合產生聚合物,同時產物發生一定程度的消旋化。因此,硫化反應的較佳溫度為65 ℃。
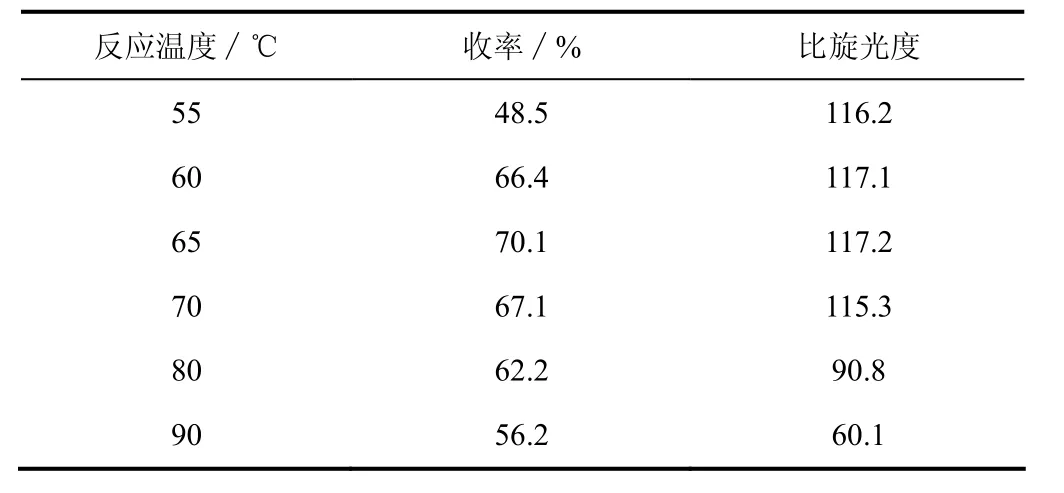
表3 溫度對硫化反應的影響
2.3.2 相轉移催化劑用量對硫化反應的影響
以50 mL無水乙醇為溶劑,在投料比n[R-(+)-6,8-二氯辛酸乙酯]∶n(Na2S2)=1∶1,硫化反應溫度65 ℃、反應時間6 h、55 ℃水解反應2 h條件下,改變相轉移催化劑四丁基溴化銨的用量,考察其對R-(+)-α-硫辛酸收率及比旋光度的影響,結果見表4。
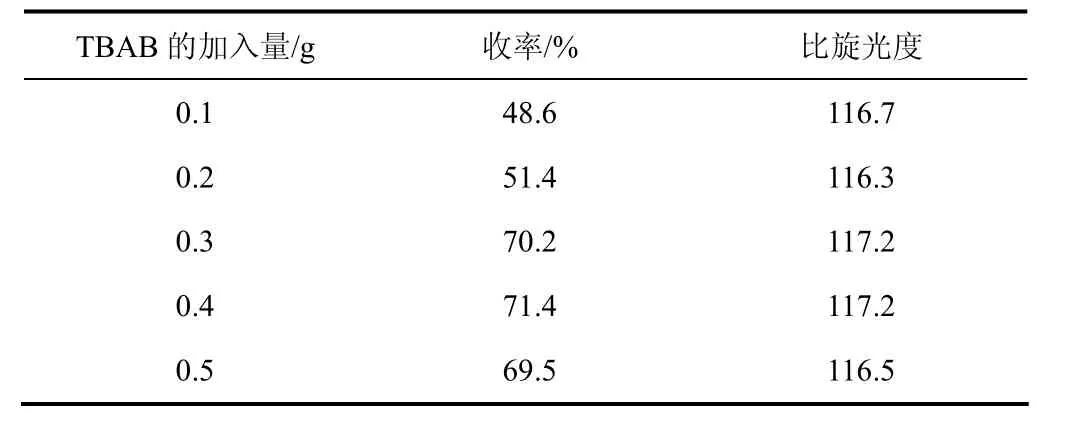
表4 相轉移催化劑(TBAB)用量對硫化反應的影響
由表4可知,當相轉移催化劑用量較小時,反應收率較低,隨著相轉移催化劑TBAB用量的增加,反應收率不斷提高,這是因為TBAB的(C4H9)4N+與水相中的S2-結合形成鹽,使得S2-更容易與R-(+)-6,8-二氯辛酸乙酯中的Cl-發生親核取代,所以收率會提高。當用量超過0.4 g時收率開始降低,是由于TBAB過量,致使大量的S2-同時進攻R-(+)-6,8-二氯辛酸乙酯的6位和8位C原子,雙硫鍵彼此產生聚合,生成了副產物,因此相轉移催化劑的用量取0.4 g為最佳。
2.4 硫化反應機理的探討
R-(+)-6,8-二氯辛酸乙酯與Na2S2發生的取代反應為合成R-(+)-α-硫辛酸的關鍵步驟,由于R-(+)-6,8-二氯辛酸乙酯中6位和8位碳上的雜原子Cl有未共享的電子對,使得其α-碳原子更容易被進攻,6位α-碳原子具有位阻效應,因此,其反應歷程可以分兩步:第一步,雙硫基團的一端在8位碳原子上發生取代反應;第二步,另一端在6位碳原子上發生取代并且分子內環化[19]。反應歷程如圖4所示。
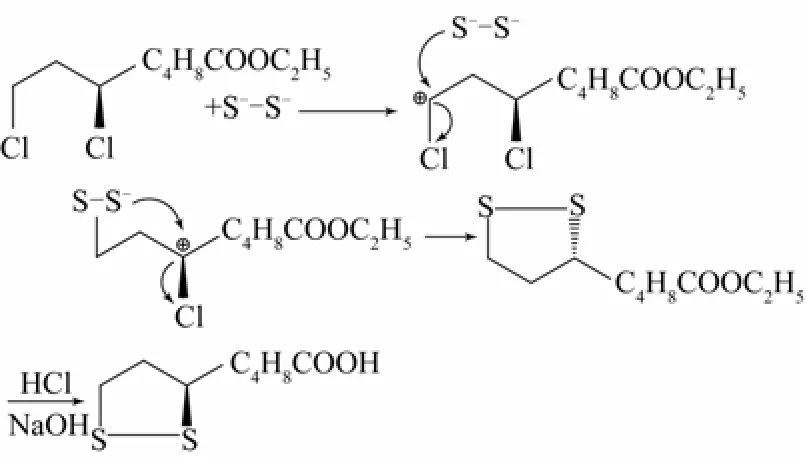
圖4 R-(+)-6,8-二氯辛酸乙酯的硫化反應機理
實驗證明,硫化反應得到的R-(+)-α-硫辛酸旋光性較好,這就排除了SN1機理,按照SN1機理,親核試劑分別從兩邊進攻碳正離子,兩種結合方式分別生成了構型保持和構型轉化產物,得到的是外消旋產物。因此,推測此反應為SN2機理,即雙硫基團一端的S-先從背面進攻位阻較小的8位碳,成鍵的同時造成Cl-離去。此時另一端沒有成鍵的S-繼續進攻手性6位碳,Cl-離去,構型翻轉,形成了含有2個S雜原子的五元環,旋光性仍然為右旋。
3 結 論
(1)用S-(-)-α-苯乙胺拆分原料6,8-二氯辛酸,得到R-(+)-6,8-二氯辛酸;經酯化制得R-(+)-6,8-二氯辛酸乙酯;最后經硫化水解合成R-(+)-α-硫辛酸。以6,8-二氯辛酸計三步總收率為44.3%,高于其它文獻值。拆分后的S-(-)-6,8-二氯辛酸經消旋化可再利用,此法條件溫和,易于工業化推廣。
(2)合成R-(+)-6,8-二氯辛酸的較佳條件:投料比為n[S-(-)-α-苯乙胺]∶n(6,8-二氯辛酸)= 0.45∶1,溶劑為乙酸乙酯,純度97.0%,收率72.8%;合成R-(+)-6,8-二氯辛酸乙酯的較佳條件:催化劑為對甲苯磺酸,反應時間7 h,純度96.4%,收率93.7%;合成R-(+)-α-硫辛酸的較佳條件:硫化溫度65 ℃,相轉移催化劑用量0.4 g,純度97.3%,收率71.4%。
[1] Packer L,Kraemer K,Rimbach G. Molecular aspects of lipoic acid in the prevention of diabetes complication[J]. Nutrition,2001,17:888-895.
[2] 徐兆瑜. 維生素E的替代品——硫辛酸[J]. 精細與專用化學品,2000(16):15.
[3] Samantha B,Pirngruber G D,Roel P. Influence of the properties of zeolite BEA on its performance in the nitration of toluene and nitrotoluene[J]. J. Catal.,2004:297-303.
[4] Wincenty S Monika M. Nitration of toluene with 65% nitric acid over MoO3/SiO2as catalyst[J]. Przem. Chem.,2002,81(8):519-521.
[5] 田部浩三,小野嘉夫,等著. 鄭祿彬,等譯. 新固體酸和堿及其催化作用[M]. 北京:化學工業出版社,1992:64-65.
[6] Tanabe K. Application of niobium oxides as catalysis[J]. Catal. Today,1990(8):1-11.
[7] Golding B T,Brookes M H,Howes D A. Method for producing lipoic acid and dihydrolipoic acid[J]. J. Chem. Soc.,1983,19:1051-1053.
[8] Elliott J D,Steele J,Johnson W S. An efficient asymmetric synthesis of (R)-(+)-α-lipoic acid [J]. Tetrahedron Letters,1985,26(21):2535-2538.
[9] Sudalai A,Upadhya T T,NikalajeM D,et al. A symmetric dihydroxylation and hydrogenation approaches to the enantioselective synthesis of R-(+)-α-lipoic acid [J]. Tetrahedron Letters,2001,42(29):4891-4893.
[10] Gopalan A S,Jacobs H K.Bakers yeast reduction of alkyl 6-chloro-3-oxohexanoates:Synthesis of (R)-(+)-α-lipoic acid [J]. Journal of the Chemical Society,Perkin Trans.,1990,7:1897-1900.
[11] 韓麗,楊倩,孫志浩,等. 脂肪酶催化拆分外消旋6-羥基-8-氯辛酸乙酯[J]. 精細化工,2007,24(6):573-576.
[12] 曾松,狄延鑫,宋寶安,等.(R)-α-硫辛酸合成方法的研究進展[J]. 精細化工中間體,2007,37(5):1-5.
[13] 楊芹,冷一欣,林富榮,等. α-硫辛酸及其R型異構體的合成[J]. 江蘇工業學院學報,2009,21(4):20-23.
[14] Flavio V P,Antonio N P D,Annibale F M,et al. Synthesis of R-(+)-α-lipoic acid:US,2004/0002610 A1[P]. 2004-01-01.
[15] Chavan S,Shivsankar K,Pasupathy K. Simplistic expedient and practical synthesis of (±)-α-lipoic acid [J]. Synthesis,2005,8(5):1297-1300.
[16] Chavan S,Praveen C,Ramakrishna G,et al. Enantioselective synthesis of R-(+)-α and S-(-)-α-lipoicacid [J]. Tetrahedron Letters,2004,45(31):6027-6028.
[17] 孫賢祥,高曉新,孫凌志. 非對映體結晶拆分中的溶劑效應[J]. 常州大學學報:自然科學版,2010,22(4):69-74.
[18] 賴卡特 C著. 唐培楚等譯. 有機化學中的溶劑效應[M]. 北京:化學工業出版社,1987:175-176.
[19] 花文廷. 朱新郵譯. 雜環化學[M]. 北京:北京大學出版社,1991:20-24.
Synthesis of R-(+)-α-lipoic acid
LENG Yixin,NIU Jinsen,HUANG Chunxiang
(School of Petrochemical Engineering,Changzhou University,Changzhou 213164,Jiangsu,China)
R-(+)-6,8-dichlorooctanic acid was obtained in chiral resolution of 6,8-dichlorooctanic acid by using chiral split agent S-(-)-α-phenylethylamine. R-(+)-α-lipoic acid was synthesized by sulfuration and hydrolysis from R-(+)-6,8-dichlorooctanic acid ethyl ester that was prepared through esterfication reaction. The conditions were investigated and the results showed that the yield and specific rotation were influenced by feed ratio,solvent,catalyst and reaction temperature. Proper experimental condition for chiral reaction was found as:n[S-(-)-α-phenylethylamine]∶n(6,8-dichlorooctanic acid)=0.45∶1,solvent was ethyl acetate. The optimal esterfication reaction conditions was found as:the catalyst was p-toluene sulfonic acid,reaction time 7 h. The optimum conditions of sulfuration reaction were found as:reaction temperature was 65 ℃,the amount of phase transfer catalyst was 0.4 g; The overall yield was 48.4%. The product and intermediate were characterized by IR,specific rotation and1H NMR.
6,8-dichlorooctanic acid;chiral resolution;esterfication;sulfuration;R-(+)-α-lipoic acid
O 626.2
A
1000-6613(2012)06-1325-05
2011-12-07;修改稿日期:2012-02-16。
及聯系人:冷一欣(1961—),女,博士,教授,研究方向為綠色化學品和醫藥中間體的合成。E-mail rxslyxcn@yahoo.com.cn。
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
