丙烯醛/氨反應制備3-甲基吡啶的研究進展
2012-10-19 03:36:12晁自勝黃登高羅才武王開明潘金鋼
化工進展 2012年5期
關鍵詞:催化劑
張 弦,晁自勝,黃登高,羅才武,劉 偉,王開明,潘金鋼
(1湖南大學化學化工學院化學生物傳感與計量學國家重點實驗室,湖南 長沙 410012;2青島科技大學化學與分子工程學院,山東 青島 266042;3北京華地博源生化科技有限公司,北京 100120)
進展與述評
丙烯醛/氨反應制備3-甲基吡啶的研究進展
張 弦1,晁自勝1,黃登高1,羅才武1,劉 偉1,王開明2,潘金鋼3
(1湖南大學化學化工學院化學生物傳感與計量學國家重點實驗室,湖南 長沙 410012;2青島科技大學化學與分子工程學院,山東 青島 266042;3北京華地博源生化科技有限公司,北京 100120)
綜述了丙烯醛/氨反應制備3-甲基吡啶的方法,主要包括液相釜式反應法、氣相固定床反應法和氣相流化床反應法3種。介紹了這些方法的工藝特點,評述了其優缺點及所涉及的催化劑。對丙烯醛/氨反應制備3-甲基吡啶過程所需要著重解決的問題進行了歸納總結。并簡介了3-甲基吡啶的合成機理。同時,對于丙烯醛/氨反應制備 3-甲基吡啶技術的發展前景也進行了展望,認為介孔材料和固體酸催化劑應用于該反應及合成機理的深入研究是未來的發展方向之一。
3-甲基吡啶;丙烯醛;液相法;氣相法
3-甲基吡啶是制備B族維生素、煙酸和煙酰胺的主要原料,還可以用來合成新型農藥、香料及醫藥中間體[1-3]。目前在我國主要由少數幾家外資或合資企業采用壟斷技術進行生產,其3-甲基吡啶產品主要用于內部下游生產和出口,無法提供給國內其它企業,而我國每年需要大量進口3-甲基吡啶。隨著3-甲基吡啶應用途徑的不斷拓展,產品缺口將不斷加大。另外,目前工業上主要采用甲醛/乙醛法[4]來生產3-甲基吡啶,此方法的產物中 4-甲基吡啶的含量較高(3-甲基吡啶/4-甲基吡啶為0.9~2.8)。由于3-甲基吡啶與4-甲基吡啶的物理化學性質極為接近(沸點相差0.5 ℃),采用常規方法難以將二者分離,從而導致分離成本增加、產品純度低[5]。而采用丙烯醛為原料制備 3-甲基吡啶時產物不含4-甲基吡啶,可以滿足各種使用要求,大大降低了分離費用。此外,目前在工業上丙烯醛是通過丙烯部分氧化進行大規模生產的[6],并且通過丙烷選擇性氧化[7]及甘油選擇性脫水[8-9]合成丙烯醛的技術也正在不斷完善。原料丙烯醛來源廣泛而穩定,且價格較低,因而深入了解丙烯醛制備3-甲基吡啶的工藝及技術具有重要工業及市場價值。同時,相關研究也能給其它雜環化合物的制備帶來借鑒作用。
1 3-甲基吡啶的合成概況
3-甲基吡啶合成方法很多。最早是由煤焦油和焦爐氣中分離而得來的,由于產物復雜,產量有限,無法滿足生產要求[10]。Akhmerov等[11]采用乙烯/甲醇/氨(摩爾比)=1/1/1、在420 ℃反應得到14.5%的3-甲基吡啶。由于乙烯轉化率不高,產物收率低,難以工業化應用。二甲基戊二腈(或胺)[12-14]是直接合成3-甲基吡啶的另一途徑。Lanini等[14]考察了2-甲基戊二腈制備3-甲基吡啶的催化劑、選擇性以及操作條件對反應的影響。通過六碳有機物的脫氫(或加氫)、成環可以得到 70%左右的3-甲基吡啶收率,主要缺點在于原料難以得到,催化劑需要用到貴金屬,價格昂貴。此外,Sreekumar等[15]利用吡啶和甲醇制備3-甲基吡啶,選擇性達到 95%以上,主要存在轉化率不高、原料成本較高的問題。
工業上3-甲基吡啶是在乙醛和氨合成2-甲基吡啶和4-甲基吡啶工藝的基礎上加入甲醛合成的。采用HZSM-5(90)為催化劑,乙醛/甲醛/氨=1/1/4(摩爾比)為原料,在375 ℃進行反應,吡啶收率57%,3-甲基吡啶收率為 27.0%[16]。改變原料的組成,可調節產物的組成比例,因而可以根據市場需求隨時調整產物。該工藝反應溫度高,存在碳損失導致大量吡啶生成,同時會減少催化劑壽命,需采用流化床來實現連續生產。另外,3-甲基吡啶中會存在較多的4-甲基吡啶,在分離過程中會損失部分3-甲基吡啶,限制了3-甲基吡啶的生產。由于吡啶堿工業的發展,工業上對3-甲基吡啶質量要求越來越高,解決甲醛/乙醛法產物含4-甲基吡啶而難以分離的問題在目前顯得極為迫切。
文獻報道丙烯醛是甲醛/乙醛法制備3-甲基吡啶的中間體[17],同時,甲醛和乙醛經過縮合可直接用來制備丙烯醛[18]。因而可以直接用丙烯醛來合成3-甲基吡啶,避免部分乙醛和氨反應直接縮合形成2-甲基吡啶和4-甲基吡啶[19],也就不需再分離3-甲基吡啶和4-甲基吡啶。該法可以用來合成高質量3-甲基吡啶,且原料價廉易得。
2 丙烯醛制備3-甲基吡啶方法
目前采用丙烯醛為原料制備3-甲基吡啶主要有液相釜式法、氣相固定床法和氣相流化床法。
2.1 液相釜式法
歐洲專利1240928[20]報道采用丙烯醛和乙酸銨為原料,在酸性條件下形成3-甲基吡啶。在250 m L丙酸溶劑中加入0.38 mol乙酸銨后,緩慢滴加0.1 mol丙烯醛,130 ℃反應得到0.0165 mol的3-甲基吡啶,其收率達到 33%。采用加入乙醛或三聚乙醛為原料和加入乙酸銅為催化劑的方法收率也無法提高。該法反應簡單、操作容易、反應溫度低、吡啶堿產物單一。但是,總吡啶堿收率不高,由于丙酸和3-甲基吡啶沸點相近,使得分離量大、分離困難。
美國專利4421921[21]報道,將2.05 mol丙烯醛原料加入到1.14 L含3.4 mol磷酸氫二銨的水溶液中,在230 ℃、3.2~3.3 MPa壓力和攪拌條件下,保溫反應10 m in。通過二氯甲烷3次萃取提取吡啶堿,得到61.9%的吡啶堿,其中3-甲基吡啶52.4%。采用部分丙烯醛用甲醛和三聚乙醛取代,加入丙烯醛1.12 mol,丙烯醛/三聚乙醛/甲醛(摩爾比)=1/1.5/1。反應得到89.8%收率的吡啶堿,其中3-甲基吡啶收率56.7%。此方法所得總吡啶堿收率高,原料利用率高,主要產物為3-甲基吡啶和3-乙基吡啶,但是反應壓力高、產物復雜、反應操作和分離提純困難。
本文作者課題組在歐洲專利1240928[20]方法的基礎上采用乙酸為溶劑,乙酸銨為氨源,以固體超強酸為催化劑在125 ℃左右反應可以得到60%左右的3-甲基吡啶,但是乙酸分離量大,難以工業化。以己酸為溶劑和磷酸銨為氨源、固體超強酸為催化劑、質量分數為8%左右的丙烯醛為原料,采用釜式反應器在170 ℃反應得到55%左右的3-甲基吡啶[22],有效提高了3-甲基吡啶反應收率,且分離簡單,不存在乙酰胺副產物,為液相法工業化提供了重要依據,但是需要解決生產能力小的問題。
總之,采用液相釜式法制備3-甲基吡啶具有反應溫度低,操作簡單和產物不含4-甲基吡啶的優點。但是也存在著分離量大和生產能力小等問題。選擇合適的溶劑和氨源,并提高3-甲基吡啶的選擇性是液相法制備3-甲基吡啶的關鍵。
2.2 氣相固定床法
以氣相固定床反應制備3-甲基吡啶主要有兩種方式:第一種是直接以純丙烯醛為原料反應制備3-甲基吡啶;另一種是采用丙烯醛和其它有機物為原料和氨反應制備3-甲基吡啶。
2.2.1 純丙烯醛制備3-甲基吡啶
歐洲專利 1222971[23]和 1208291[24]報道采用丙烯醛、氨氣、空氣和氮氣的混合氣體,丙烯醛體積分數一般維持在2%~5%,氨為7%~20%,在反應溫度為400 ℃左右、接觸時間2~5 s的條件下,分別采用HF或(和)Zr改性SiO2-A l2O3和硼酸改性SiO2為催化劑,總吡啶堿的收率最高可達68%左右。由于氧的存在,主要產物為吡啶,3-甲基吡啶收率只有25%左右。氧會導致部分丙烯醛氧化而損失,同時吡啶堿收率降低。由于氧化劑和水的存在,能及時將催化劑上積炭氧化,保持催化劑較長時間的活性,因而,反應可以維持較長時間。
日本專利 5626546[25]、歐洲專利 1422601[26]以及美國專利3898177[27]、3917542[28]、3960766[29]報道以丙烯醛和氨氣反應,采用 SiO2-A l2O3和MFx/Al2O3等,在380~450 ℃條件下反應可以得到70%左右的吡啶堿收率,其中3-甲基吡啶收率可高達45%左右。反應采用高比表面積的Al2O3為催化劑,通過氧化硅、HF或者B、P、Mg、Ca等改性,可以進一步提高催化劑性能,提高吡啶堿收率。且反應必須注意讓丙烯醛和氨分別預熱至 200 ℃以上,并在反應床內混合,否則,在沒有催化劑而有氨的條件下丙烯醛可聚合,堵塞進料管道,導致反應無法進行。美國專利3898177[27]報道,采用1 L Mg和F改性的A l2O3為催化劑,5.04 mol丙烯醛和13.8 mol氮氣預熱至220 ℃后與10 mol預熱至220 ℃的氨氣在反應管內混合反應。用水洗反應氣體,并用苯(或二氯甲烷[28])萃取產物,得到總吡啶堿收率為71%,其中吡啶為26.6%,3-甲基吡啶為44%。反應24 h后催化劑活性下降,通入空氣1 h于500℃再生,催化劑活性恢復。由于催化劑積炭嚴重、壽命短,因而反應一段時間后,需要將催化劑再生。否則,催化劑失活后,丙烯醛轉化率和3-甲基吡啶選擇性降低,未反應丙烯醛在氨和水作用下聚合堵塞管道。為了保證丙烯醛轉化率,需丙烯醛過量,因此氨醛比一般為1~3,氨不宜太過量,否則容易導致丙烯醛聚合,降低3-甲基吡啶選擇性。丙烯醛的空速一般為0.8~2.5 s。空速過快,會降低丙烯醛轉化率,而未反應丙烯醛又會聚合導致管道堵塞;空速過慢,容易使丙烯醛過度脫羰,催化劑積炭嚴重。為了減少丙烯醛的聚合,一般通入惰性氣體(或溶劑)如氮氣、水蒸氣或苯等稀釋,然后與丙烯醛混合一起預熱,之后再與預熱后的氨在催化床層接觸反應。
高溫氣相反應容易導致丙烯醛脫羰積炭,因此固定床反應催化劑需要壽命長、再生性能好。歐洲專利1422601[26]使用直徑2 mm、長度4~6mm、比表面積300 m2/g的氧化鋁為載體,采用浸漬法加入Mg、Zr和Ba鹽等及氟化物為活性組分,干燥后于700 ℃空氣氛中焙燒 4 h。催化劑原子比A l/Mg/Ti/F=1000/25/25/100,丙烯醛/氨/氮(摩爾比)=10/19.5/20.4,可以得到313 g/ h每升催化劑的產率,總吡啶堿收率為 71%,其中 3-甲基吡啶為46.4%,吡啶為24.6%。美國專利3917542[28]和美國專利3960766[29]同樣報道采用F改性氧化鋁為催化劑,并加入B、P、Mg、Zr和Ti等作為輔助成分改性,同樣得到70%左右的吡啶堿,3-甲基吡啶收率在 30%~40%。文獻[30]報道采用二氧化鈦為催化劑,證實丙烯醛和氨反應合成3-甲基吡啶反應過程3-甲基吡啶選擇性的提高主要是由于催化劑 Lew is酸中心催化引起的。
該法由于積炭嚴重,導致吡啶收率較高,從而降低3-甲基吡啶收率,且催化劑反應一段時間需要再生,導致操作復雜,從而要求催化劑壽命長、再生性能好。
2.2.2 丙烯醛和其它有機物制備3-甲基吡啶
為了解決采用純烯醛為原料時存在催化劑失活快的問題,特別是裂解反應導致吡啶生成、3-甲基吡啶減少的問題,考慮添加其它有機物為原料,減少丙烯醛損失,減緩催化劑失活,提高3-甲基吡啶收率。所加有機物主要是醛、酮、醇和環氧丙烷。
歐洲專利963887[31]、887688[32]采用丙烯醛和飽和醛與氨氣 350~450 ℃反應制備吡啶堿。當采用丙烯醛和乙醛[31]時,得到產物吡啶堿收率最高可達65%左右,其中3-甲基吡啶只有26.5%,吡啶為28%,產物2-甲基吡啶和4-甲基吡啶分別為8.5%,對產物分離帶來較大困難,也影響到了產品質量,同時催化劑壽命不能得到較大的提高。提高丙烯醛和乙醛的比例,可以適當提高產物3-甲基吡啶和吡啶的比例,但比例過大,會導致總吡啶堿收率降低。向混合氣體中加入氧氣和水蒸氣,能夠提高催化劑壽命、減少結焦,但是導致3-甲基吡啶減少,吡啶增多,并降低總的吡啶堿收率。而采用丙烯醛和丙醛[32]反應,丙醛/丙烯醛(摩爾比)=(1/1)~(1.2/1),氨/醛(摩爾比)=(0.5/1)~(5/1),反應溫度為375~475 ℃,采用 SiO2-Al2O3為催化劑得到 50%左右的 3-甲基吡啶轉化率(基于總醛),產物吡啶堿不含4-甲基吡啶。3-甲基吡啶收率明顯提高,可見,丙醛可以參與3-甲基吡啶的形成。可能源于丙醛脫氫與丙烯醛反應得到的反應中間體,在有氨條件下形成3-甲基吡啶。
歐洲專利 896049[33]報道采用丙烯醛/丙醇(摩爾比)=14.8/1,氨/碳(摩爾比)=1.1/1為碳源,原料經氣化后在400 ℃反應,3-甲基吡啶選擇性只有31.4%(基于丙烯醛或丙醇),明顯低于丙烯醛和丙醛反應所得。可見丙醇反應活性不如丙醛,考慮丙醇參與合成3-甲基吡啶的反應過程需要脫去兩分子氫,反應更困難,可能需要加強催化劑氧化性能或通入一定量氧參與反應。另外,將原來丙烯醛和丙醇一起蒸發時,二者容易發生縮合形成縮醛,導致3-甲基吡啶收率降低,應將二者分別預熱后在反應器內再混合反應為宜。歐洲專利920526[34]采用丙烯醛和酮為原料,在400 ℃反應,得到最高18%左右的 3-甲基吡啶轉化率(基于丙烯醛),產物隨所添加酮的不同而有較大的變化。當加入丙酮和丙烯醛與氨反應時,產物主要為2-甲基吡啶和3-甲基吡啶,其中3-甲基吡啶轉化率只有25%(基于丙烯醛),大大降低了3-甲基吡啶收率,且另外生成了2-甲基吡啶和3-甲基吡啶,使產物復雜,增加分離成本。歐洲專利1192255[35]采用丙烯醛/環氧丙烷/氨氣(摩爾比)=5/1/9,接觸時間為3 s,反應溫度400~450 ℃,得到48.6%的3-甲基吡啶和12.6%的吡啶收率。國內山東化學研究所[36-37]采用氟改性的 A l2O3催化劑,以丙烯醛和環氧丙烷反應得到14%的吡啶和57%的3-甲基吡啶收率,總吡啶堿收率進一步提高,達到72%左右;并提出HO-A lF1~4為該催化劑的活性中心,且催化活性一定范圍內隨表面酸度增加而增加,但是反應時間不長,經過一段時間后,催化劑活性下降,容易出現聚合物堵塞反應管道。
在固定床中進行反應,丙烯醛和氨在接觸催化劑之前應該分別預熱到 200~240 ℃左右,而且最好直接在反應管內混合[28]。丙烯醛可以用氮氣或苯蒸氣稀釋,產物3-甲基吡啶也要用溶劑萃取回收。由于反應催化劑容易積炭,壽命較短,因此應及時檢測反應收率變化情形,及時將催化劑再生,否則,催化劑失活后未反應丙烯醛容易聚合,堵塞反應管道。同時,為了延長催化劑壽命,必要時需要稍微改性降低催化劑活性,以減少催化劑再生次數,因此,選用氧化鋁比表面積一般在200~350 m2/g。反應產物也應用水或其它溶劑吸收,并用苯或二氯甲烷等有機溶劑萃取分離,并及時蒸發分離。在所添加有機物中,加入醇、酮反應所得3-甲基吡啶收率不是很高。環氧丙烷和丙醛效果最好,產物主要為3-甲基吡啶,且收率可以達到60%左右。環氧丙烷和丙醛形成3-甲基吡啶的過程中,可以通過高溫脫氫,直接參與形成3-甲基吡啶,提高3-甲基吡啶收率。且脫氫在一定程度上還可以還原催化劑,減少脫碳反應的進行,減緩積炭程度,延長催化劑壽命。
2.3 氣相流化床法
采用流化床可以使失活的催化劑迅速離開反應區,避免因催化劑失活帶來的丙烯醛轉化率降低,丙烯醛和氨氣聚合堵塞反應器出口的問題。同時,為了適當減少丙烯醛自聚,考慮加入適量醛或酮,特別是丙醛,在提高3-甲基吡啶收率,避免4-甲基吡啶生成的同時,其它吡啶也很少,具有產物單一、分離成本低等優點。
歐洲專利 1020857[38]報道采用丙烯醛/水蒸氣/氨氣(摩爾比)=1/1/5,反應溫度350 ℃,使用B、P改性的比表面積為600 m2/g的SiO2-A l2O3為催化劑,接觸時間5 s。在流化床上吡啶選擇性為25%,3-甲基吡啶為35%。經過17 h反應后催化劑積炭達到18%,催化劑活性下降。美國專利4171445[39]報道,在反應器-再生器雙塔裝置上由丙烯醛和氨氣合成吡啶堿,可以得到24.2%吡啶和48.5%的3-甲基吡啶。丙烯醛氣化后與氮氣混合在反應器底部作為一路進料,氨氣和氮氣在催化劑下,反應器下部側線作為一路進料。尾氣經過催化劑床后,用水洗回收吡啶堿產物,未反應的氮氣和氨氣循環作原料使用。在反應器內設計有一定的自由空間,供催化劑移動至再生器,在再生器內經過空氣氧化再生后回到反應器內,因此,可以保持催化劑的高活性,提高3-甲基吡啶收率,并保證反應連續運行。
歐洲專利 1069368[40]報道采用體積分數為5.1%的丙烯醛、5%的乙醛、34.8%的氮氣、8.7%的氧和18.4%的水蒸氣為原料,用Pb和F改性的SiO2-A l2O3為催化劑,在溫度400 ℃、接觸時間為5.5 s條件下使用流化床反應器反應7 h,得到吡啶收率為 46%、3-甲基吡啶和 4-甲基吡啶混合物為 4%左右、2-甲基吡啶為 1%。可見由于氧的存在導致主要產物為吡啶,且由于乙醛含量較高,導致 2-甲基吡啶和 4-甲基吡啶出現。加拿大專利1063121[41]采用丙烯醛和丙酮,反應溫度為400~450 ℃,催化劑為高比表面的硅酸鋁,在流化床上得到 30%左右的 3-甲基吡啶和 25%左右的 2-甲基吡啶。美國專利4147874[42]、4163854[43]報道采用丙烯醛分別和乙醛、丙醛作為原料反應,以高比表面積SiO2-A l2O3為催化劑,在 400~460 ℃之間反應。采用丙烯醛/乙醛=2/1的原料,得到46%左右的3-甲基吡啶收率和 27%左右的吡啶收率。采用丙烯醛/丙醛=2/1的原料時,得到 60.6%的 3-甲基吡啶和6.2%的吡啶收率。改變丙烯醛/丙醛摩爾比在3/2~4/1之間,吡啶和3-甲基吡啶收率變化不大,吡啶收率維持在 6%~10%,3-甲基吡啶收率維持在60%左右。可見采用丙烯醛和丙醛為原料,3-甲基吡啶收率較高,可達60%,總吡啶堿收率接近 70%,且產物不含 4-甲基吡啶,達到較好的反應結果。
綜上所述,丙烯醛和氨反應可以制備高純度3-甲基吡啶,總結各種合成方法,其優缺點如表 1所示。采用流化床可以使反應連續進行,且3-甲基吡啶收率可達60%,總的吡啶堿可以達70%左右。主要存在投資成本高、風險大、操作復雜等問題。
3 關鍵技術及存在問題
3.1 丙烯醛的聚合
丙烯醛的化學性質極為活潑,在光、熱、酸和堿的作用下都可以發生聚合反應。通常將對苯二酚加入丙烯醛作為阻聚劑,并用乙酸調節pH=5~6,配制乙酸/對苯二酚/無水碳酸鈉=84/8/8(質量比)的緩沖溶液抑制丙烯醛自聚[44]。
文獻[45]報道在光引發作用下氣態丙烯醛可以聚合形成固體顆粒。Ohgomori等[46]報道丙烯醛通過尾-尾聚合形成2-烯基己二醛。同時由于丙烯醛的兩個不飽和鍵共軛,兩鍵可以圍繞 C—C鍵旋轉,因而存在順式和反式兩種結構,使得丙烯醛的聚合更加復雜化。Toma等[47]報道丙烯醛通過Diels-A lder反應二聚形成兩種不同結構的二聚丙烯醛。
丙烯醛由于同時含有C=C鍵和C=O鍵,不僅存在自聚,還可以和其它不飽和物質發生縮聚或共聚。文獻[48]報道了丙烯醛、苯乙烯和二乙烯苯的共聚。Yamashita等[49-50]報道了丙烯醛在不同溶劑條件下的聚合,并認為丙烯醛主要通過陰離子引發聚合。

表1 丙烯醛制備3-甲基吡啶方法比較
丙烯醛容易自聚,在加熱狀態下,不需催化劑就可以生成二聚丙烯醛,因此需加入阻聚劑和 pH值緩沖劑,可采用在反應之前預熱或通入氮氣稀釋丙烯醛以減少自聚。而氨氣高溫下活性增強,極易引發丙烯醛的聚合,因此,不能讓丙烯醛和氨氣混合后再加熱,只能分別預熱后在催化劑床上直接接觸反應,且預熱溫度不得低于200 ℃[51]。但是即便如此,只是減少了聚合反應時間,減少聚合程度,無法完全避免高聚物產生。在反應物料中加入添加劑也能起到一定的阻聚作用,如水蒸氣、醛、酮等[34-37]。由于丙烯醛聚合能力較強,因此反應過程必須保證轉化率達 100%,否則,未反應的丙烯醛在氨水的作用下聚合成酯,堵塞反應管道。為了防止丙烯醛自聚,在反應之前加入大量的稀釋氣或添加劑,又會阻礙丙烯醛的反應轉化率。目前的主要辦法是加入吸收劑和萃取劑處理反應產物。美國專利 3898177[27]采用水吸收反應產物,并加入苯萃取蒸餾反應產物,得到71%左右的吡啶堿收率。美國專利 3917542[28]報道采用水洗和二氯甲烷萃取蒸餾的辦法同樣得到70%左右的吡啶堿收率。美國專利4237299[52]報道采用兩個清洗塔-熱交換裝置來回收以丙烯醛和乙醛為原料反應得到吡啶堿產物。第一個填充柱清洗塔裝置將產物清洗并降溫至 40~80 ℃,第二個塔裝置將余下產物進一步降溫至 10~40 ℃。降溫后液體產物經苯萃取后,精餾得到吡啶和3-甲基吡啶,回收率分別達到98.4%和99.5%。該裝置回收效果明顯,避免了反應管道的堵塞問題。
總之,通過預熱、隔離、稀釋和吸收的辦法,可以大大降低丙烯醛的聚合,關鍵在于維持丙烯醛的高轉化率。同時采取高效的吸收裝置處理反應產物,避免堵塞現象。
3.2 丙烯醛的裂解
在高溫下,該反應容易脫碳形成吡啶產物,其中吡啶/3-甲基吡啶達到1/2左右,造成催化劑壽命的降低和丙烯醛損失加大。目前報道的主要方法有通入水蒸氣[23]、通氧[24]、采用高效催化劑[27-29]和流化床反應器[39]等辦法。通氧和水蒸氣可以有效地使沉積炭減少、延長催化劑使用壽命。但通氧會使部分原料氧化,使產物總吡啶堿收率降低,吡啶增加的同時而3-甲基吡啶大大減少。水蒸氣的加入可以使吡啶和3-甲基吡啶收率同時提高,且總吡啶堿收率達到62%左右,但水的加入降低了反應平衡同時使分離成本增加。添加其它有機物可以有效提高總收率,其中以丙醛[43]、丙酮[34,41]和環氧丙烷[35-37]效果較佳,在不產生4-甲基吡啶的基礎上提高了3-甲基吡啶收率,同時產生高沸點的其它烷基吡啶堿,但并不能避免催化劑壽命的降低。通過提高催化劑載體比表面積[27-29]、酸處理[21]和載體強度[27]等辦法都可以延長催化劑的使用壽命,提高總收率。但總的催化劑壽命有限,必須通過不斷再生使催化劑重復使用才可實現工業化目的。流化床反應器裝配有兩段反應器[39],其中一個反應器反應,另一反應器再生,催化劑在兩個反應器內不斷循環往復使用,解決了催化劑積炭嚴重、壽命較短的問題。但需要催化劑強度高、重復性好,同時反應裝置和操作費用會增加。
通過加入有機物的方法可以在丙烯醛裂解的基礎上大幅提高3-甲基吡啶收率,但催化劑壽命問題主要通過再生的辦法來解決。因此合適的反應類型應是低溫液相反應或流化床反應,流化床反應則需要強度更高、再生性能更好的催化劑。
3.3 催化劑
丙烯醛和氨反應制備3-甲基吡啶從反應平衡來講,不涉及氧化及還原,因而,不宜用氧化性或還原性催化劑。主要的反應類型有加成、縮合、脫水(氨)、成環和氫轉移等,因而酸性或堿性催化劑較為適宜。
液相反應[20]中酸性溶劑就起到一定的催化作用,然而酸性又不宜太強,否則,引起丙烯醛聚合,無法得到3-甲基吡啶;且酸性太強,難以釋放氨,導致反應無法進行,同時酸會腐蝕設備、污染環境等。因而液相反應采用有機酸為溶劑或采用緩沖溶液,調節并穩定 pH值,使反應體系為弱酸性。開發新型固體酸催化劑取代液體酸或鹽催化劑,既能避免腐蝕問題,又能提高反應收率,具有重要意義。
氣相反應主要使用的催化劑有 SiO2[24]、TiO2[30]、A l2O3[26-29,36-37]、Al2O3-SiO2[22-23,53]和分子篩[54]等,這類催化劑的主要特征是催化劑具有一定的酸堿性,且比表面積高(300~800 m2/g),有利于縮合、脫水反應的進行。為了進一步提高3-甲基吡啶的選擇性,可以加入Mg、Ca、Ba等增強催化劑活性[28-29]。另外,通過含F、B、P元素的化合物處理催化劑可以進一步調節并改善催化劑的酸堿性質[23-24,36-37]。在流化床反應中,為了提高催化劑的耐磨性和穩定性,可以適當加入一定量的Zr或Ti組分[26-27]。Zenkovets等[30]認為Lew is酸中心為主要的催化劑活性中心。
丙烯醛合成3-甲基吡啶對催化劑的要求根據反應工藝的不同會有所不同,根據工藝選擇合適的催化劑至關重要。同時,隨著新的固體酸堿催化劑的不斷涌現以及新的催化劑改進技術的應用,特別是介孔材料催化劑有望在此反應中發揮重要作用。同時,催化劑相關催化機理還需進一步深入探索。
4 丙烯醛合成3-甲基吡啶機理
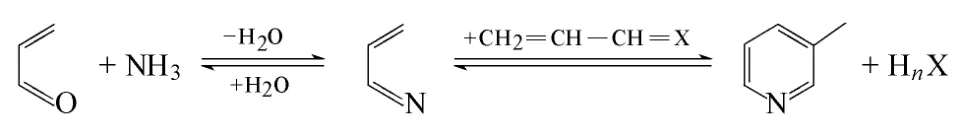
圖1 丙烯醛制備3-甲基吡啶形成機理
目前,尚無丙烯醛/氨反應制備3-甲基吡啶的機理報道,不過在甲醛/乙醛法合成 3-甲基吡啶過程中,丙烯醛被認為是反應中間體。根據醛/氨反應合成吡啶堿相關機理報道[55],丙烯醛和氨的反應一般認為是先形成丙烯亞胺(CH2=CH—CH=NH)中間體,然后,一分子丙烯亞胺與一分子丙烯醛或者丙烯亞胺縮聚形成3-甲基吡啶,如圖1所示。
然而,丙烯醛在低溫和高溫下反應均可形成3-甲基吡啶,不同溫度下反應機理是否相同還有待進一步研究。由丙烯亞胺形成3-甲基吡啶不涉及脫氫反應,只存在脫水縮合的過程,但會涉及加成、脫水(脫氨)、氫轉移和成環等步驟,各步驟之間如何協調轉化,且丙烯醛法和甲醛/乙醛法在合成3-甲基吡啶機理過程是否一致還需詳細論證。
5 結 語
丙烯醛法制備3-甲基吡啶是一種合成高質量3-甲基吡啶的有效方法,該反應原料來源穩定,產品質量較高。目前主要存在的問題是原料容易聚合、催化劑壽命短、價格較甲醛/乙醛法高等。隨著丙烯醛來源的不斷拓展,其價格有望降低,該法將更具競爭力。但是目前,此法的相關專利技術掌握在國外公司手中,隨著我國對3-甲基吡啶需求的加大,加快該技術領域的研究,形成自己的獨立知識產權具有重要戰略意義。
致謝 感謝北京華地博源生化科技有限公司和中國海洋科技大學李國強教授對本課題的支持!
[1] 呂金魁,魏榮寶,梁婭,等. 新型煙用香料3-乙基吡啶的合成[J].天津理工學院學報,1995,11(2):71-75.
[2] Martin A,Janke C,Kalevaru V N. Ammoxidation of 3-picoline to nicotinonitrile over VPO catalysts[J].Applied Catalysis A:General, 2010,376(1–2):13-18.
[3] 溫占興,霍竹林,焦素霞. 3-甲基吡啶法生產2-氯-5-三氟甲基吡啶[J]. 河北化工,2000(1):10-13.
[4] Jin Fang,Cui Yugang,Rui Zebao,et al. Effect of sequential desilication and dealum ination on catalytic performance of ZSM-5 catalyst for pyridine and 3-picoline synthesis[J].Journal of Materials Research,2010,25(2):271-282.
[5] 趙歡,肖國民. 3-甲基吡啶和4-甲基吡啶的分離技術進展[J]. 化工科技,2004,12(4):52-56.
[6] 王樂夫,耿建銘,徐建昌,等. 丙烯選擇性氧化制丙烯醛得催化劑研究[J]. 高校化學工程學報,1999,13(4):377-381.
[7] 王鑒,李安蓮,邴國強,等. MoVTeNbO催化劑用于丙烷選擇氧化與氨氧化研究進展[J]. 化工進展,2010,29(12):2298-2303.
[8] Hanan Atia,Udo Armbrusterb,Andreas Martin. Dehydration of glycerol in gas phase using heteropolyacid catalysts as active compounds[J].Journal of Catalysis,2008,258:71-82.
[9] 張躍,丁海亮,劉建武,等. H3PW12O40/Al2O3催化劑在甘油脫水制備丙烯醛反應中的性能評價[J]. 化工進展,2010,29(2):261-265.
[10] 常景泉. 魯奇氣化煤焦油中重吡啶堿類的提取與精制[J]. 煤化工,2004,32(6):39-41.
[11] Akhmerov K M,Yusupov D,Abdurakhmanov A,et al. Catalytic synthesis of pyridine and methylpyridine from acetylene and ammonia[J].Khimiya Geterotsiklicheskikh Soedinenii,1975(2):221-224.
[12] Lanini S,Prins R. Synthesis of 3-picoline from 2-methylglutaronitrile over supported noble metal catalysts I. Catalyst activity and selectivity[J].Applied Catalysis A:General,1996,137:287-306.
[13] Prins R. Synthesis of picolines and other aza-aromatics from arylam ines by isomerization-rearrangement and from dinitriles by hydrogenation-cyclization reactions[J].Catalysis Today,1997,37:103-120.
[14] Lanini S,Prins R. Synthesis of 3-picoline from 2-methylglutaronitrile over supported noble metal catalysts II. Influence of operation parameters[J].Applied Catalysis A:General,1996,137:307-326.
[15] Sreekumar K,Mathew Thomas,Devassy Biju M,et al. Vapor-phase methylation of pyridine w ith methanol to 3-picoline over Zn1-xCoxFe2O4(x=0,0.2,0.5,0.8 and 1.0)-type ternary spinels preparedviaa low temperature method[J].Applied Catalysis A:General,2001,205:11-18.
[16] Kuppi Reddy Suresh Kumar Reddy,Inkollu Sreedhar,Kondapuram Vijaya Raghavan. Interrelationship of process parameters in vapor phase pyridine synthesis[J].Applied Catalysis A:General,2008,339(1):15-20.
[17] Sato Hiroshi,Shimizu Shinkichi,Abe Nobuyuki,et al. Synthesis of pyridine bases over ion-exchange pentasil zeolite[J].Chemisty Letters,1994,23(1):59-62.
[18] Dum itriu E,Bilba N,Lupascu M,et al. Vapor-phase condensation of formaldehyde and acetaldehyde into acrolein over zeolites[J].Journal of Catalysis,1994,147(1):133-139.
[19] 楊風玉,史玉龍,丁春年,等. 2-甲基吡啶和4-甲基吡啶合成工藝研究[J]. 化工時刊,2004,18(11):41-43.
[20] Asrian Nicolson. Manufacture of pyridine bases:GB,1240928[P]. 1971-07-28.
[21] James I Grayson,Rolf Dinkel. Process for the production of 3-picoline:US,4421921[P]. 1983-12-20.
[22] 張弦,晁自勝,李國強,等. 一種利用丙烯醛制備 3-甲基吡啶的新方法:中國,102249989[P]. 2011-11-23.
[23] Graham Sw ift,Ian Stuart M ccoll. Catalytic process for the manufacture of pyridine or methylpyridines:GB,1222971[P]. 1971-02-17.
[24] Palph Hubert Benson,Peter Arthur Edward Whincup. Catalytic process for the manufacture of pyridine and methylpyridines:GB,1208291[P]. 1970-10-14.
[25] Kiyoshi Yasuda,Kazuyuki Matsuoka. Catalyst for manufacturing pyridine bases:JP,5626546[P]. 1981-03-14.
[26] Hans Schaefeer,Helmut Bescke,Gerd Schreyer,et al. Catalyst the production of pyridine and 3-methylpyridine:GB,1422601[P]. 1976-01-28.
[27] Helmut Beschke,Hans Schaefer,Gerd Schreyer. Catalysts for the production of pyridine and 3-methylpyridine:US,3898177[P]. 1975-08-05.
[28] Helmut Beschke,Axel Kleemann,Gerd Schreyer. Catalysis for the production of pyridine and 3-methylpyridine:US,3917542[P]. 1975-11-04.
[29] Helmut Beschke,Franz-ludw ing Dahm,Friedrich,et al. Catalyst for the production of pyridine and 3-methylpyridine:US,3960766[P]. 1976-06-01.
[30] Zenkovets G A,Volodin A M,Bedilo A F,et al. Influence of the preparation procedure on the acidity of titanium dioxide and its catalytic properties in the reaction of synthesis ofβ-picoline by condensation of acrolein w ith ammonia[J].Kinetics and Catalysis,1997,38(5):669-672.
[31] Kenneth Raymond Hargrave. Production of pyridine andβ-picoline:GB,963887[P]. 1964-07-15.
[32] Kenneth Raymond Hargreave. Production ofβ-picoline:GB,887688[P]. 1962-01-24.
[33] Kenneth Raymond Hargreave. Production ofβ-picoline:GB,896049[P]. 1962-05-09.
[34] Kenneth Raymond Hargreave. Production of pyridine bases:GB,920526[P]. 1963-03-06.
[35] Yoshiaki Numa,Akio Nakamachi,Yasukazu Murakam i. Process for producing pyridine andβ-picoline:GB,1192255[P]. 1970-05-20.
[36] 王彩彬,李玉潤. 由丙烯醛合成β-甲基吡啶的研究[J]. 中國醫藥工業雜志,1984(6):1-5.
[37] 王彩彬,李玉潤. 經氟處理的氧化鋁在合成β-甲基吡啶中的催化作用及活性中心結構[J]. 催化學報,1982,3(3):187-191.
[38] Ian Campbell,John Anthony Corran. Manufacture of pyridine and 3-mehylpyridine:GB,1020857[P]. 1966-02-23.
[39] Helmut Beschke,Heinz Friedrich,Gerd Schreyer,et al. Process for the production of pyridine and 3-methyl pyridine:US,4171445[P]. 1979-10-16.
[40] Antony Harold Patrick Hall. Production of pyridine and picolines:GB,1069368[P]. 1967-05-17.
[41] Helmut Beschke. Process for the production of 2-methylpyridine and 3-methyl pyridine:CA,1063121[P]. 1979-09-25.
[42] Helmut Beschke,Heinz Friedrich. Process for the production of pyridine and 3-methyl pyridine:US,4147874[P]. 1979-04-03.
[43] Helmut Beschke,Heinz Friedrich. Process for the production of 3-methyl pyridine:US,4163854[P]. 1979-08-07.
[44] 張旭之,陶志華,王松漢,等. 丙烯衍生物工學[M]. 北京:化學工業出版社,1995:391-398.
[45] Morita Hiroshi,Semba Katsuhiko,Umezawa Takeshi,et al. Photochem ical fine particle formation in the gas phase from acrolein by a two-photon process[J].Colloids and Surfaces A:Physiochemical and Engineering Aspects,1999,153:203-207.
[46] Ohgomori Yuji,Ichikawa Shuji,Sumitani Naoko. Tail-to-tail dimerization reaction of acrolein[J].Organometallics,1994,13:3758-3760.
[47] Toma Lucio,Quadrelli Paolo,Caramella Pierluigi. Classical and non-classical secondary orbital interactions and coulombic attraction in the regiospecific dimerization of acrolein[J].Tetrahedron Letters Pergamon,2001,42:731-733.
[48] Zanio G Scampini,A lcino P De Aguiar,M?nica R M P Aguiar,et al. Oxime groups introduction in copolymer networks based on acrolein[J].Materials Letters,2004,58:3933-3938.
[49] Yamashita Natsuki,Yoshihara Masakuni,Maeshima Toshihisa. Polymerization of acrolein in the presence of acrolein induced by pyridine and water system[J].Polymer Letters,1972,10:643-646.
[50] Yamashita Natsuki, Inoue Hiroshi, Maeshima Toshihisa. Polymerization of acrolein and methyl vinyl ketone induced by am ine-water and pyridine-phenol systems[J].Journal of Polymer Science:Polymer Chemistry Edition,1979,17:2739-2747.
[51] Graham Sw ift. Catalytic process for the manufacture of pyridine and methylpyridine:GB,1158365[P]. 1969-07-16.
[52] Helmut Beschke,Franz-ludw ing Dahm,Friedrich,et al. Process for the recovery of pyridine and 3-methylpyridine: US,4237299[P].1980-12-02.
[53] Ivanova A S,Al’kaeva E M,Mastikhin V M,et al. Physicochem ical and catalytic properties of silica-alum ina catalysts in the reaction of acrolein condensation w ith ammonia[J].Kinetics and Catalysis,1996,37(3):425-430.
[54] Parks-Sm ith David G. Manufacture of pyridine and 3-methylpyridine:CA,895885[P]. 1972-03-21.
[55] Calvin J R,Davis R D, M cAteer C H. Mechanistic investigation of the catalyzed vapor-phase formation of pyridine and quinoline bases using13CH2O,13CH3OH,and deuterium-labeled aldehydes[J].Applied Catalysis A:General,2005,285:1-23.
Advances in the synthesis of 3-picolineviaacrolein/ammonia reaction
ZHANG Xian1,CHAO Zisheng1,HUANG Denggao1,LUO Caiwu1,LIU Wei1,WANG Kaiming2,PAN Jingang3
(1State Key Laboratory of Chemo/Biosensing and Chemometrics,College of Chemisty and Chemical Engineering,Hunan University,Changsha 410012,Hunan,China;2College of Chem istry and Molecular Engineering,Qingdao University of Science and Technology,Qingdao 266042,Shandong,China;3Beijing Huadi Boyuan Biochem ical Technology Lim ited Company,Beijing 100120,China)
Abatract:This paper reviewed the synthesis of 3-picolineviathe reaction between acrolein and ammonia. It was reported up to now that there are mainly three approaches for the synthesis of 3-picoline from the reaction of acrolein w ith ammonia,i.e.,the liquid-phase reaction in stirring tank reactor,the gas-phase reaction in fixed-bed reactor and the gas-phase reaction in fluidized-bed reactor. The paper described the general features of these reaction processes,w ith comments on their advantages and disadvantages and the related catalysts. It addressed particularly the key issues in the reaction process of acrolein/ammonia to 3-picoline. The foreground of the mesoporous material and solid acid catalysts applied to the reaction and the mechanism researches of 3-picoline prepared from acrolein were also prospected,the mesoporous material and solid acid catalysts applied to the reaction and the mechanism researches were thought to one of the development direction.
3-picoline;acrolein;liquid-phase method;vapor-phase method
TQ 253.21
A
1000–6613(2012)05–1113–08
2011-11-12;修改稿日期:2012-02-05。
湖南省芙蓉學者獎勵計劃。
張弦(1983—),男,博士研究生。聯系人:晁自勝,教授,博士生導師。E-mail zschao@hnu.edu. cn。
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
