二苯醚水解耦合苯酚氧化羰基化合成碳酸二苯酯的熱力學分析
2012-11-09 02:47:34杜治平林志坤黃麗明吳元欣
石油化工 2012年8期
杜治平,林志坤,黃麗明,袁 華,吳元欣
(武漢工程大學 綠色化工過程教育部重點實驗室
湖北省新型反應器與綠色化學工藝重點實驗室,湖北 武漢 430073)
二苯醚水解耦合苯酚氧化羰基化合成碳酸二苯酯的熱力學分析
杜治平,林志坤,黃麗明,袁 華,吳元欣
(武漢工程大學 綠色化工過程教育部重點實驗室
湖北省新型反應器與綠色化學工藝重點實驗室,湖北 武漢 430073)
以碳酸二甲酯和碳酸二乙酯的熱力學數據為基礎,結合Benson、Joback和馬沛生基團貢獻法估算了碳酸二苯酯(DPC)的熱力學性質,進而計算了苯酚氧化羰基化合成DPC反應和二苯醚水解反應的焓變、熵變、吉布斯自由能變和平衡常數。計算結果表明,兩個反應均為放熱反應;在5 MPa、353~413 K下,兩個反應均為自發過程,其中苯酚氧化羰基化反應的平衡常數較大,且隨溫度的升高而降低;二苯醚水解反應的平衡常數較小,且隨溫度的升高而增大。為達到耦合除水的目的,兩個反應的反應速率需維持合適的比例。
碳酸二苯酯; 苯酚;氧化羰基化; 二苯醚; 水解; 耦合;熱力學分析
碳酸二苯酯(DPC)是一種重要的有機化工原料,其合成方法有光氣法、碳酸二甲酯酯交換法、草酸二苯酯脫羰法和苯酚氧化羰基化法等[1-4]。苯酚氧化羰基化法是以CO、O2和苯酚為原料一步合成DPC,它具有原子利用率高、理論上水是惟一的副產物和無污染等優點,但副產物水對催化劑有毒害作用,催化劑的活性較低且壽命較短;另外,水也能促進DPC水解。當前主要采用分子篩脫水[5-6],但分子篩脫水存在如下缺陷:(1)分子篩自身的吸水能力有限,而在反應溫度(一般為353 K)下分子篩的吸水能力會因升溫而進一步削弱,脫水效果不理想;(2)分子篩表面的酸、堿中心對DPC的水解有促進作用。因此必須探尋新的脫水方法。
苯酚由于酚羥基的碳氧鍵比較牢固,一般不能通過苯酚的分子間直接脫水制備二苯醚(DPE),而是通過酚鈉與鹵代芳烴在Cu的催化作用下,經高溫反應合成。由于DPE的合成較為困難,因此經水解生成苯酚的過程可能更為容易。因此,能否通過二苯醚的原位水解以消耗苯酚氧化羰基化反應中生成的水,在不引入雜質的情況下,實現水解反應與羰基化反應的耦合,從而提高DPC的合成收率。
本工作從熱力學的角度對DPE水解和苯酚氧化羰基化反應進行了熱力學計算和分析,以期對實驗研究提供理論支持。
1 反應原理和熱力學計算
1.1 反應原理
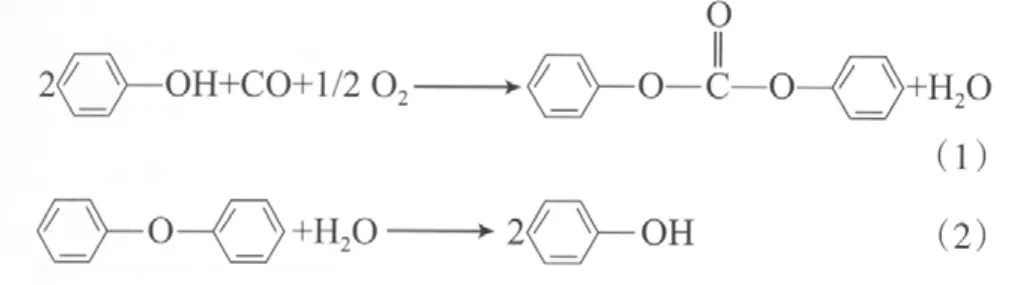
苯酚氧化羰基化和DPE水解反應如下:用—OCOC—的氣態定壓熱容替代—OCOO—的氣態定壓熱容等[7],這樣很難保證數據的可靠性。因此,本工作以文獻報道的相關碳酸酯的熱力學數據為基礎,通過計算—OCOO—的相關熱力學數據,進而求解DPC的相關熱力學性質。
1.2.1 298.15 K下氣態DPC的熱力學數據
1.2.1.1 氣態DPC的標準摩爾生成焓和標準摩爾熵
298.15 K下,晶體DPC的標準摩爾生成焓和標準摩爾熵分別為-401.66 kJ/mol和278.60 J/(mol·K)[8],而相同溫度下DPC的升華焓為90 kJ/mol[9],因此,由相平衡可計算298.15 K下,氣態DPC的標準摩爾生成焓和標準摩爾熵分別為-311.66 kJ/mol和580.46 J/(mol·K)。
1.2.1.2 氣態DPC的摩爾定壓熱容
由于缺少DPC的摩爾定壓熱容數據,而Benson基團貢獻法考慮了臨近基團的影響[10],具有較高的精度,因此采用Benson基團貢獻法估算DPC的摩爾定壓熱容。計算公式見式(3)。

1.2 熱力學計算
關于DPC熱力學數據的計算已見報道,但由于缺少碳酸酯基(—OCOO—)的相應數據,多采用類似—OCOO—的其他基團數據進行替代,如采
由于缺少—OCOO—的定壓熱容數據,因此,由碳酸二乙酯(DEC)的摩爾定壓熱容[11],采用Benson基團貢獻法求取,進而估算DPC的相應熱力學參數,估算結果見表1。

表1 DEC和DPC的基團貢獻值Table 1 Thermodynamic data of group contribution of diethyl carbonate(DEC) and diphenyl carbonate(DPC)
根據表1中的數據,進一步通過線性回歸得到DPC的摩爾定壓熱容與溫度(T)的關系,關系式見式(4)。
Cpm=-43.267+1 006.300×10-3T-500.000×10-6T2(4)
1.2.1.3 DPC的臨界溫度和沸點下的蒸發焓
依據Joback對基團的劃分原則[12],并根據DEC的臨界溫度(575.98 K)和沸點(400.00 K)[11]計算 —OCOO—的基團貢獻值(見表2),進而根據DPC的沸點(574.7 K)估算出DPC的臨界溫度為794.16 K。計算公式見式(5)。
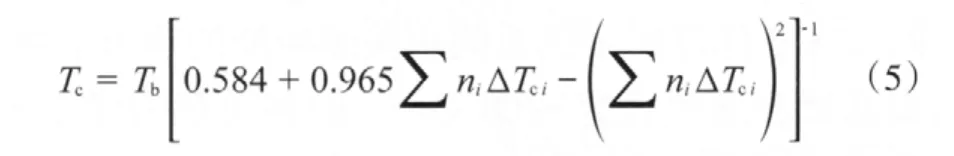
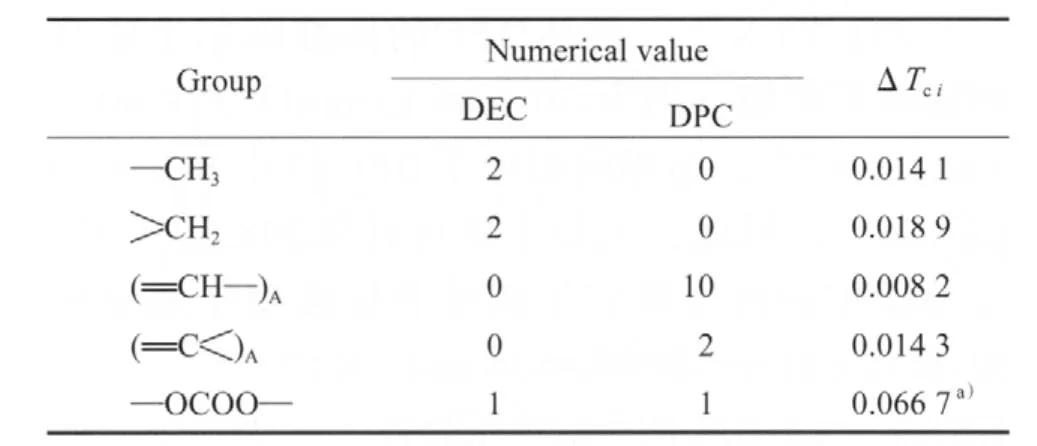
表2 Joback法估算Tc的基團貢獻值Table 2 Group contribution values of Tc calculated based on Jaback method
苯酚氧化羰基化反應為液相反應,但缺少DPC的蒸發焓數據。由于馬沛生基團貢獻法采用了雙官能團劃分法[13],相比常用的單官能團,Joback和Reid基團貢獻法具有更高的準確度,因此采用該法計算DPC的蒸發焓。計算公式見式(6)。
由于缺少完整的—OCOO—的貢獻值,所以選取沸點下的DMC蒸發焓(33.82 kJ/mol)[14]和DEC蒸發焓(36.31 kJ/mol)[11],計算—OCOO—沸點下的基團貢獻值(見表3),進而求得DPC沸點下的蒸發焓為51.88 kJ/mol。
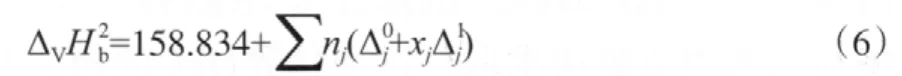

表3 馬沛生基團貢獻法估算ΔVHb的基團貢獻值Table 3 The group contribution value of calculating ΔVHb based on the Ma Peisheng method
1.2.2 各組分的蒸發焓
由各組分的沸點、臨界溫度和沸點下的蒸發焓(見表4),采用Watson公式[15](式(7))估算它們在不同溫度下的蒸發焓,估算結果見表5。

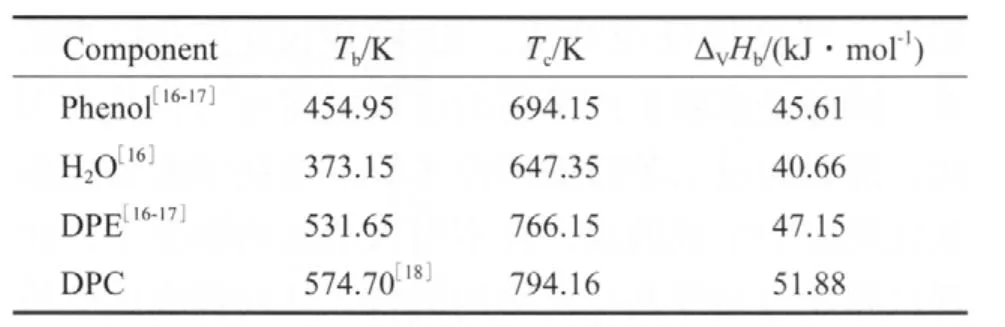
表4 各組分的沸點、臨界溫度和沸點下的蒸發焓Table 4 Tb,Tc and ΔVHb of different components

表5 各組分在不同溫度下的蒸發焓Table 5 The vaporization enthalpies of the components at different temperatures
1.2.3 各組分的液體生成焓
根據各組分在不同溫度下的氣態摩爾定壓熱容(見表6),用式(8)計算各組分的氣態摩爾生成焓,計算結果見表7。

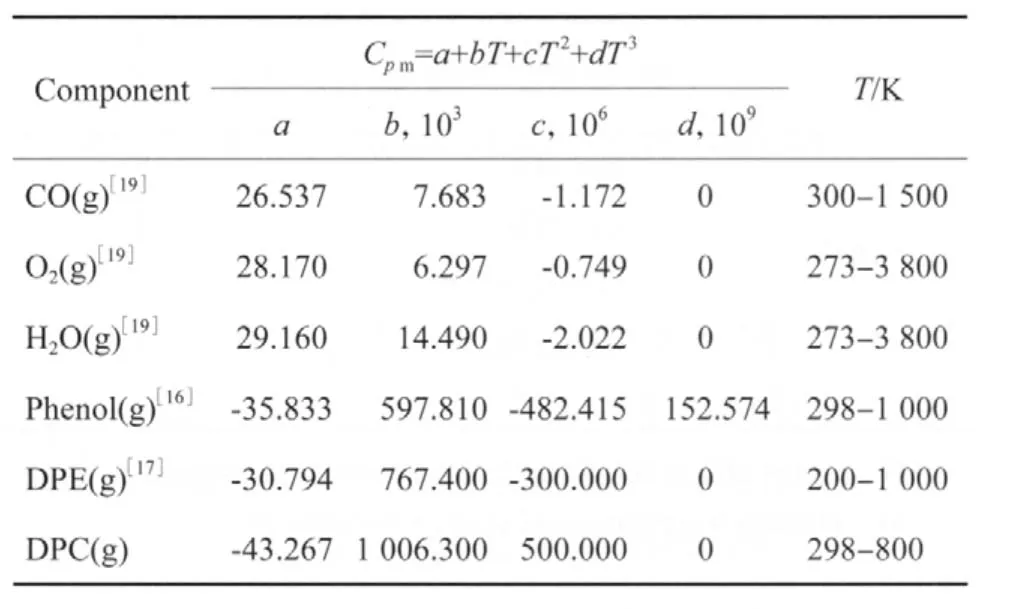
表6 不同溫度下各組分的氣態摩爾定壓熱容Table 6 The gaseous molar heat capacity at constant pressure of different components at different temperatures
根據各組分在不同溫度下的氣態摩爾生成焓和摩爾蒸發焓,用式(9)計算各組分的液態摩爾生成焓,計算結果見表7。


表7 各組分在不同溫度下的氣態和液態標準摩爾生成焓Table 7 The gaseous and liquid molar formation enthalpy of the components at different temperatures
1.2.4 各組分的液態標準摩爾熵
根據理想氣體等壓變化時的熵變計算公式(式(10)),計算不同溫度下氣態物質的標準摩爾熵,再根據該物質發生相變時的摩爾蒸發焓,用式(11)計算該物質的液態標準摩爾熵,計算結果見表8。
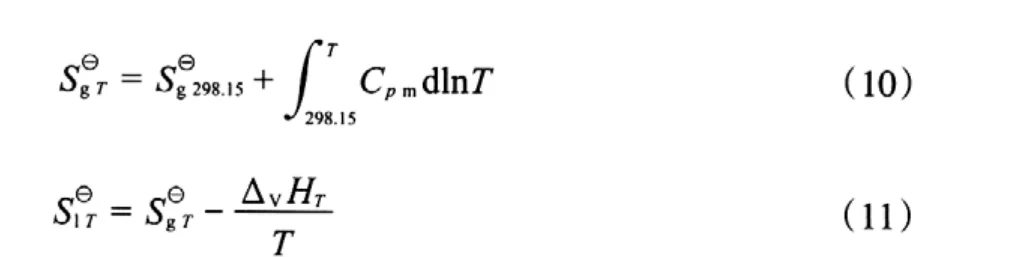
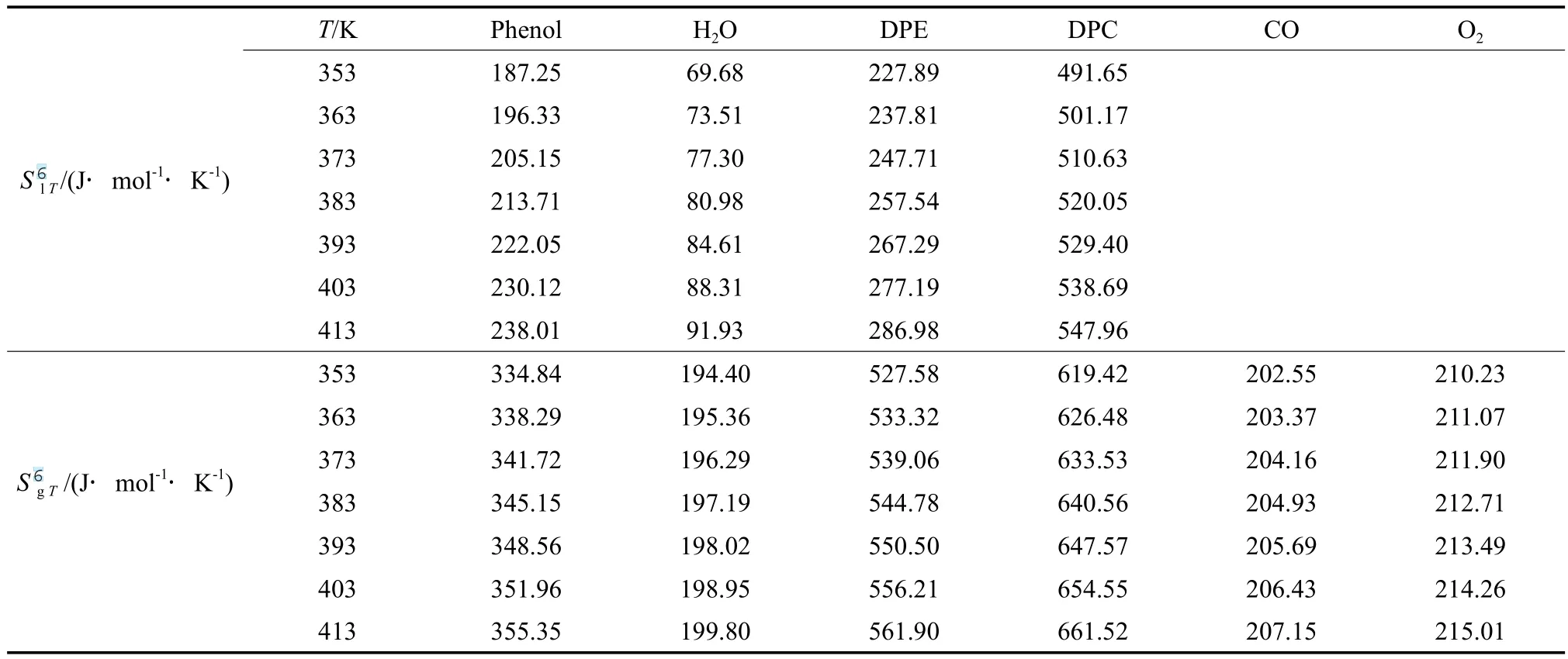
表8 各組分在不同溫度下的氣態和液態標準摩爾生成熵Table 8 The gaseous and liquid molar formation entropy of the components at different temperatures
2 結果與討論
苯酚氧化羰基化反應為分子數減少的反應,為促進DPC的生成,一般需要加壓。根據文獻[4]報道的結果,選取典型工藝條件為壓力5 MPa、溫度353~413 K進行計算。根據反應中各組分所處的狀態,分別設計了如圖1所示的狀態變化過程計算苯酚氧化羰基化反應和二苯醚水解反應的焓變(ΔrHm)、反應的吉布斯自由能變(ΔrGm)、反應的熵變(ΔrSm)和反應平衡常數(K6)(忽略各組分混合以及壓力變化對液體的影響)。

圖1 苯酚氧化羰基化反應和二苯醚水解反應的狀態設計Fig.1 State design of the oxidative carbonylation of phenol and the hydrolysis of DPE.
2.1 反應焓變的計算
2.1.1 等溫變壓過程(S1→S2)ΔH1的計算
苯酚氧化羰基化反應中,苯酚為液態,忽略壓力對其焓的影響;同時,CO和O2近似為理想氣體,由于反應過程的溫度不變,所以該過程的焓變近似為0。二苯醚水解反應中,二苯醚、水和苯酚均為液態,壓力對其焓的影響可忽略,所以該過程的焓變也可近似為0。即ΔH1≈0。
2.1.2 標準狀態下反應過程(S2→S3)ΔrH62的計算
根據表7中的數據,用式(12)計算標準狀態下苯酚氧化羰基化反應和二苯醚水解反應的ΔrH62。

2.1.3 等溫變壓過程(S3→S4)ΔH3的計算
反應過程中DPC、苯酚和水均為液態,忽略壓力對其焓的影響,所以該過程的焓變近似為0。即ΔH3≈0。
2.1.4 不同溫度下反應過程(S1→S4)ΔrHm的計算
在5 MPa下,用式(13)計算不同溫度下苯酚氧化羰基化反應和二苯醚水解反應的ΔrHm,計算結果見表9。從表9可知,苯酚氧化羰基化反應和二苯醚水解反應的焓變均為負值,表明兩個反應都是放熱反應,其中前者的熱效應較大,在所計算的溫度范圍內放熱量均在255 kJ/mol以上,且隨溫度的變化較小;后者呈現微弱的放熱效應,且隨溫度的升高放熱量逐漸減小。在所計算的溫度范圍內,二苯醚水解反應的焓變與苯酚氧化羰基化反應的焓變的比值在0.16%~0.56%之間,因此,從熱力學角度來看,當兩反應耦合進行時,二苯醚水解反應對苯酚氧化羰基化反應的影響很小。

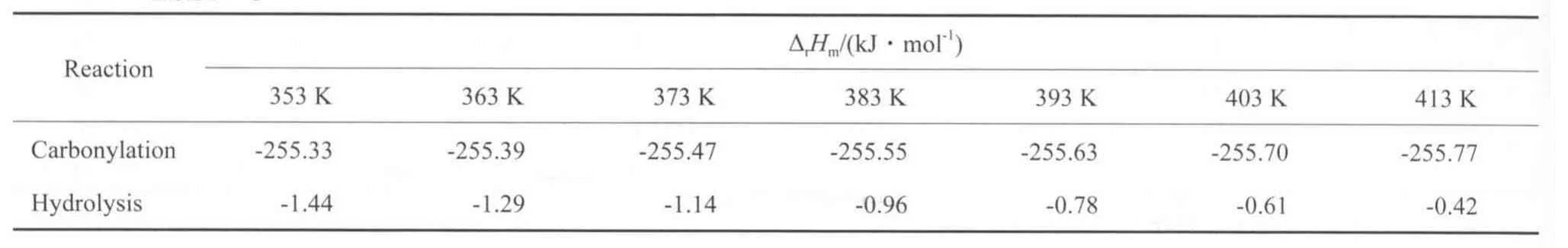
表9 不同溫度下苯酚氧化羰基化反應和二苯醚水解反應的焓變Table 9 The enthalpy changes in the carbonylation of phenol and the hydrolysis of diphenyl ether at different temperatures
2.2 反應吉布斯自由能變的計算
忽略壓力對液相組分熵的影響,用式(14)計算氣相組分的熵變,用式(15)計算標準狀態下的反應熵變ΔS6,用式(16)計算5 MPa下的反應熵變
r2ΔrSm。結合ΔrHm和ΔrSm,用式(17)計算5 MPa下的反應吉布斯自由能變ΔrGm,計算結果見表10。
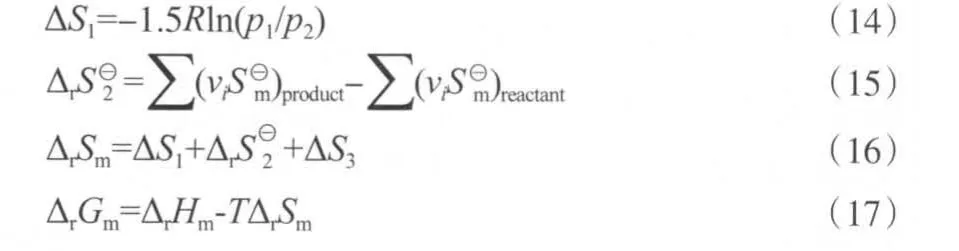
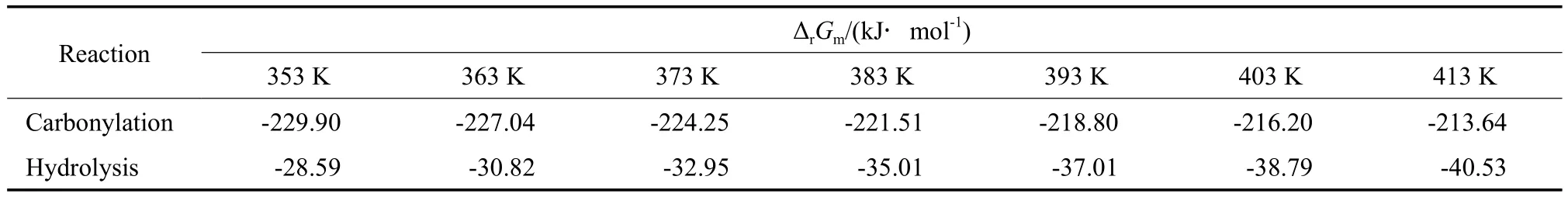
表10 不同溫度下苯酚氧化羰基化反應和二苯醚水解反應的吉布斯自由能變Table 10 The Gibbs free energy changes in the carbonylation of phenol and the hydrolysis of diphenyl ether at different temperatures
從表10可知,苯酚氧化羰基化反應和二苯醚水解反應的吉布斯自由能變均為負值,說明兩個反應在計算的溫度范圍內可以自發進行。在等溫等壓下,根據De Donder提出的反應耦合的判據[20]:(ΔrGm1)Tpr1+(ΔrGm2)Tpr2≤0(式中,下角標1和2分別代表參與耦合的兩個反應),結合表10的數據,苯酚氧化羰基化反應和二苯醚水解反應的耦合在熱力學上能夠自發進行。
2.3 反應平衡常數的計算
采用式(18)分別計算了0.1 MPa、不同溫度下的苯酚氧化羰基化反應和二苯醚水解反應的標準平衡常數(K6) ,計算結果見表11。

表11 不同溫度下苯酚氧化羰基化反應和二苯醚水解反應的平衡常數Table 11 The equilibrium constants of the carbonylation of phenol and the hydrolysis of diphenyl ether at different temperatures

從表11可知,苯酚氧化羰基化反應的平衡常數較大,說明該反應較容易進行;但由于該反應為強放熱反應,所以隨溫度的升高,平衡常數降低。從熱力學角度分析,升高溫度不利于DPC的生成;但在動力學上,當反應溫度過低時,反應速率較慢,同樣不利于DPC的合成。相比苯酚氧化羰基化反應,二苯醚水解反應的平衡常數要小得多,說明反應雖能自發進行,但反應的自發程度不如苯酚氧化羰基化反應的大。另外,二苯醚水解反應的平衡常數隨溫度的升高而增加,表明升高溫度是有利于二苯醚水解反應的進行。結合表9和表11,二苯醚水解反應的熱效應較小,基本不影響苯酚氧化羰基化反應,而苯酚氧化羰基化反應釋放的大量熱量又有利于二苯醚水解反應,因此,兩者的耦合能很好地相互促進;此外,引入二苯醚水解反應的目的在于消耗反應中的副產物水,由于苯酚氧化羰基化反應比二苯醚水解反應容易進行,如果苯酚氧化羰基化反應進行得過快,那么生成的水量將遠遠超出二苯醚水解反應所消耗的水量,則水對催化劑活性和DPC水解的影響依然存在。因此,在催化劑的設計中,要使兩個反應的反應速率維持合適的比例。
3 結論
(1)苯酚氧化羰基化反應和二苯醚水解反應均為放熱反應,且后者釋放出的熱量僅為前者的0.16%~0.56%;當兩個反應耦合時,二苯醚水解反應對苯酚氧化羰基化反應的影響很小。
(2)在5 MPa、353~413 K下,苯酚氧化羰基化反應和二苯醚水解反應的吉布斯自由能變均為負值,兩個反應均可自發進行。
(3)苯酚氧化羰基化反應的平衡常數較大,且隨溫度的升高而降低;與苯酚氧化羰基化反應相比,二苯醚水解反應的平衡常數較小,且隨溫度的升高而增大,兩個反應的耦合能很好地相互促進。
(4)為達到二苯醚水解而消耗反應中副產物水的目的,在催化劑的設計中,要使兩個反應的反應速率維持合適的比例。
符 號 說 明


[1] 馬新賓,黃守瑩,王勝平,等. 氧化羰基化法合成有機碳酸酯的研究進展[J]. 石油化工,2010,39(7):697 - 705.
[2] Du Zhiping,Xiao Yanhua,Chen Tong,et al. Catalytic Study on the Transesterification of Dimethyl Carbonate and Phenol to Diphenyl Carbonate[J].Catal Commun,2008,9(2):239 -243.
[3] 曹平,石衛兵,楊先貴,等. 酯交換法合成碳酸二苯酯的技術分析[J]. 石油化工,2010,39(3):346 - 352.
[4] Yang Xiaojun,Han Jinyu,Du Zhiping,et al. Effects of Pb Dopant on Structure and Activity of Pd/K-OMS-2 Catalysts for Heterogeneous Oxidative Carbonylation of Phenol[J].Catal Commun,2010,11(7):643 - 646.
[5] 楊小俊,吳元欣,韓金玉,等. Pd/錳氧化物催化苯酚氧化羰基化合成碳酸二苯酯:Ⅰ. 載體晶型結構和表面氧物種的影響[J]. 石油化工,2009,38(7):728 - 732.
[6] Zhao Yueqing,Liang Yinghua,Jia Qianyi,et al. Preparation of CuO-CoO-MnO/SiO2Nanocomposite Aerogels as Catalyst Carriers and Their Application in the Synthesis of Diphenyl Carbonate[J].J Wuhan Univ Technol,Mater Sci Ed,2011,26(4):595 - 599.
[7] 杜治平,王越,王公應. 碳酸乙烯酯與苯酚酯交換反應的熱力學分析[J]. 天然氣化工,2005,30(1):21 - 25.
[8] Sinke G C,Hildenbrand D L,McDonald R A,et al. The Heat,Entropy and Free Energy of Formation of Diphenyl Carbonate[J].J Phys Chem,1958,62(11):1461 - 1462.
[9] 王勝平. 苯酚和草酸二甲酯酯交換反應合成碳酸二苯酯[D].天津:天津大學,2003.
[10] Cohen N,Benson S W. Estimation of Additivity Methods,Heat Formation of Organic Compounds by Additivity Methods[J].Chem Rev,1993,93(7):2419 - 2438.
[11] 馬沛生. 石油化工基礎數據手冊:續編[M]. 北京:化學工業出版社,1993:1012 - 1013.
[12] 徐煜,衣守志,吳家全. 純物質蒸發焓估算方法的進展[J].石油化工,2009,38(7):801 - 807.
[13] 馬沛生,許文,劉貽勝,等. 用官能團法估算沸點下的蒸發焓[J]. 石油化工,1992,21(9):613 - 617.
[14] 邢愛華,張敏卿,何志敏,等. 碳酸二甲酯與苯酚酯交換合成碳酸二苯酯熱力學分析[J]. 化學工程,2006,34(11):40 - 43.
[15] 波林 B E,普勞斯尼次 J M,奧康奈爾J P. 氣液物性估算手冊[M]. 第5版. 趙紅玲,王風坤,陳圣坤,等譯. 北京:化學工業出版社,2005:45 - 182.
[16] 時鈞,汪家鼎,余國宗,等. 化學工程手冊:上冊[M]. 第2版. 北京:化學工業出版社,1996:31 - 41.
[17] 盧煥章,畢蘭云,伍章平,等. 石油化工基礎數據手冊[M].北京: 化學工業出版社,1984:584 - 585,866 - 867.
[18] 徐克勛. 精細有機化工原料及中間體手冊[M]. 北京:化學工業出版社,1998:381.
[19] 天津大學物理化學教研室. 物理化學:上冊[M]. 第4版. 北京:高等教育出版社,2002:310 - 314.
[20] Prigogine I,Delay R. Chemical Thermodynamics[M]. London:Longmans Green,1954:38 - 42.
Thermodynamic Analysis of Coupling of Oxidative Carbonylation of Phenol with Hydrolysis of Diphenyl Ether
Du Zhiping,Lin Zhikun,Huang Liming,Yuan Hua,Wu Yuanxin
(Key Laboratory for Green Chemical Process of Ministry of Education,Hubei Key Laboratory of Novel Chemical Reactor and Green Chemical Technology,Wuhan Institute of Technology,Wuhan Hubei 430073,China)
Based on the thermodynamic data of dimethyl carbonate and diethyl carbonatethe,thermodynamic properties of diphenyl carbonate were estimated by using Benson,Joback and Ma Peisheng group contribution methods,and then the enthalpy changes,entropy changes,Gibbs free energy changes and equilibrium constants in the oxidative carbonylation of phenol to diphenyl carbonate and the hydrolysis of diphenyl ether were calculated. The results show that the two reactions are exothermic and spontaneous under the conditions of 5 MPa and 353-413 K. The equilibrium constant of the oxidative carbonylation of phenol is larger than that of the hydrolysis of diphenyl ether. With the rise of temperature,the former decreases but the latter increases. It is necessary for the rates of the two reactions to maintain an appropriate proportion to remove water by the hydrolysis.
diphenyl carbonate;phenol;oxidative carbonylation;diphenyl ether;hydrolysis;coupling;thermodynamic analysis
1000 - 8144(2012)08 - 0894 - 07
TQ 225.52
A
2012 - 02 - 22;[修改稿日期]2012 - 06 - 08。
杜治平(1971—),男,湖北省仙桃市人,博士,副教授,電話 027 - 87195671,電郵 dzpxyhry@163.com。
國家自然科學基金重點項目(20936003);湖北省自然科學基金重點項目(2008CDA009)。
(編輯 安 靜)
