高脂飲食與阿爾采末病之間關系的研究進展
2012-12-06 08:03:36胡金鳳苑玉和楚世峰陳乃宏
中國藥理學通報 2012年3期
何 鑫,胡金鳳,苑玉和,楚世峰,韓 寧,陳乃宏
(1.天然藥物活性物質與功能國家重點實驗室,中國醫學科學院藥物研究所,北京 100050;2.天津中醫藥大學研究生院,天津 300193)
阿爾采末病(AD),又稱老年癡呆癥,是一種神經退行性疾病。臨床癥狀主要表現為記憶能力減退,持續性認知能力下降以及運動障礙等[1]。其主要病理特征為:顳葉和海馬皮質等部位神經元丟失;tau蛋白過度磷酸化形成神經纖維纏結(neurofibrillary tangles,NFTs);以及β淀粉樣蛋白(amyloid peptide,Aβ)大量沉積形成的老年斑(senile plaques,SPs)[2]。
AD的發病原因及病理機制錯綜復雜,不僅與遺傳因素有關,還與年齡、環境、飲食、腦外傷等密切相關。上世紀90年代就有研究報道高脂飲食是AD發病的危險因素[3]。近年來對高脂飲食與AD發病之間的相關性研究表明,中年時的高脂飲食與AD發病的危險性增加密切相關,且調節血脂的藥物如他汀類藥物、膽固醇合成阻斷劑均可防治AD[4]。腦內的膽固醇代謝紊亂與AD發病密切相關,膽固醇是調節生理狀態下脂質雙層膜物理化學狀態和功能活性的必需的分子,在中樞神經系統中不可或缺,它對包括信號傳遞、突觸可塑性、學習記憶在內的正常腦功能的維持非常重要。腦內膽固醇代謝紊亂會造成氧固醇、24-OH膽固醇、25-OH膽固醇等膽固醇代謝產物的產生發生改變,而使得腦內一些酶如乙酰基轉移酶等發生改變,進而損傷腦內神經元,造成學習記憶等的損傷及AD的發生[5]。
雖然高脂飲食可增加AD的發病率,但膽固醇不能透過血腦屏障進入腦內,所以高脂飲食并不能直接導致AD的發生。現研究表明高脂飲食引起AD發病可能與以下幾種途徑有關:①促進Aβ產生;② 誘導神經炎癥;③ 促進tau蛋白過度磷酸化;④ 損傷突觸可塑性;⑤ 損傷膽堿能神經系統。
1 高脂可促進Aβ含量增加
Aβ是老年斑的主要成分,由分泌酶裂解淀粉樣蛋白前體蛋白(APP)產生,是人體代謝的正常產物之一。流行病學調查顯示,膽固醇動態平衡的紊亂可能通過增加Aβ的蓄積而導致AD。膽固醇與Aβ聯系的最早報道是Sparks等1994在兔上實驗得到的。Jaya Prasanthi等[6]以高脂飼料飼養兔為模型來表現兔AD樣病變后大腦的病理特征。實驗表明,通過ELISA檢測表明高脂飼料飼養兔的皮層中Aβ(可溶性Aβ1-40、Aβ1-42及 Aβ 蓄積物)的含量增加,海馬中可溶性Aβ1-40、Aβ1-42含量增加,但 Aβ 蓄積物沒有增加。Ullrich等[5]研究也發現高脂飲食動物腦中Aβ的含量增加。雖然高脂飲食能導致Aβ蓄積,但其機制并不完全清楚。
已知Aβ的增加主要由兩個方面造成:生成增加以及降解受阻。Aβ在腦內的水平受裂解 APP的 β-分泌酶(BACE1)、降解Aβ的胰島素降解酶(IDE)、清除該蛋白的低密度脂蛋白相關蛋白(LRP-1)以及調節其轉運的高級糖基化終產物受體(RAGE)等因素綜合控制。BACE1是一種蛋白水解酶,在APP裂解產生Aβ的過程中起重要作用。BACE1裂解了APP的胞外區(sAPP)產生了C99膜結合C端碎片,該碎片被γ分泌酶進一步加工生成 Aβ(Fig 1)[7]。Catherine等指出高膽固醇增加 Aβ的含量是通過增加BACE1的催化活性而完成的。進一步研究發現高脂飲食可增強BACE1與其底物APP的結合,進而增加Aβ的產生。正常膽固醇條件下,BACE1分布于脂筏上,而APP在脂筏上存在的量卻很低,提示正常膽固醇條件下APP的降解方式不是通過BACE1剪切所產生。而當膽固醇含量升高,APP在細胞膜上會重新分布,到達已經包含BACE1的脂筏上,進而增加 Aβ 的生成[8]。
Aβ發揮神經損傷作用是通過作用于細胞膜表面受體,主要為RAGE、LRP-1。RAGE是一種跨膜轉運蛋白,由于其配體都具有結構特異性,而被認為是模式識別受體,其配體包括一些氧化應激產物、Aβ、APP、炎癥因子、DNA結合蛋白等[9]。LRP-1是低密度脂蛋白受體家族的一員,它擔任著多功能“清潔工”、信號分子受體、轉運體以及促使膽固醇和載脂蛋白新陳代謝的功能[10]。RAGE和LRP-1是血腦屏障(BBB)重要的毛細管運輸蛋白,在與其配體Aβ的相互作用過程中,RAGE將Aβ從血液轉運至腦中,而LRP-1能夠調節Aβ從腦到血液的轉運。因此,Aβ出腦或者入腦的凈通量依賴于這兩種受體的表達及活性,而改變這兩種蛋白的表達量及活性使其失衡則會導致Aβ轉運的不平衡,引起BBB的功能紊亂[11]。Jaya Prasanthi等[6]研究發現,高膽固醇增加海馬和皮層中RAGE的表達水平,而降低LRP-1的表達水平,使得RAGE與LRP-1的相對分布不平衡,最終導致Aβ在腦內的蓄積。
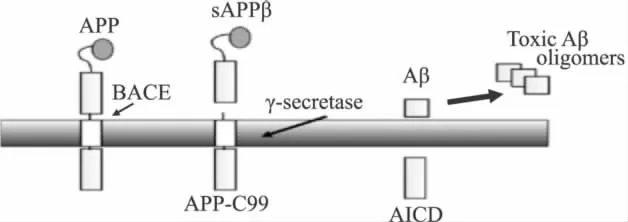
Fig 1 The releasing of Aβ from its precursor APP.
IDE是存在于胞質、過氧化物酶體、囊泡及細胞表面的一種金屬內切酶[12]。Kurochkin等[13]在1994年研究證明,IDE參與Aβ的降解。IDE是主要的Aβ降解酶,它通過對淀粉樣沉淀的組成肽的裂解而阻止其產生。IDE在皮層中能清除Aβ1-40和Aβ1-42的神經毒性作用。研究報道高脂飲食降低了海馬和皮層中IDE的表達水平,IDE的含量降低可能減弱Aβ的降解而使得 Aβ的水平升高導致腦內 Aβ的蓄積[6]。
由此可見,高脂飲食增加腦中Aβ的量與參與Aβ產生、降解、清除及轉運過程的蛋白變化有關,即(1)增強BACE1與APP的結合使Aβ的產生增加;(2)轉運Aβ由血入腦的受體RAGE含量增加,而轉運Aβ由腦入血的蛋白LRP-1含量下降,導致Aβ在腦內的蓄積;(3)Aβ降解酶IDE含量的下降,減少Aβ的清除。這些蛋白的表達變化是高脂所致Aβ蓄積的原因。因此維持正常的血漿膽固醇濃度,防止調節Aβ相關蛋白的變化,從而阻止其蓄積可防治AD。
2 引起神經炎癥
AD的關鍵性特征之一是腦內神經炎癥,而神經炎癥的主要來源是小膠質細胞的激活。激活的小膠質細胞不僅形態學上發生改變——阿米巴狀,而且在細胞內激活了NF-κB等炎性信號通路,增加誘導型一氧化氮合酶(iNOS)表達,促進NO等活性氧物質的釋放以及TNF-α、IL-1等炎性因子的分泌,間接損傷神經元[14]。Zhang等以高脂飼料飼養1個月齡的♂大鼠5個月后發現,高脂飲食可增加Iba-1的表達,激活小膠質細胞,并且明顯增加大腦皮層ROS的產生,NADPH氧化酶各亞基及COX-2的表達,增加細胞核中NF-κB的表達以及PGE2水平[15],即高脂飲食激活了腦內神經炎癥,這為高脂飲食與AD發病間的相互聯系提供了新的證據。
3 促進tau蛋白過度磷酸化
tau是一種微管相關蛋白,主要在神經細胞中表達,正常功能是誘導與促進微管蛋白合成微管,并與之結合,維持微管功能的穩定及微管蛋白分子的解離,誘導微管成束。tau的過磷酸化以及異常方式聚集可導致人類及多種動物的神經性疾病。特別是tau的過度磷酸化使其與微管蛋白的結合只有正常tau蛋白的10%,并束縛了tau蛋白對微管的穩定,造成神經纖維退化及功能喪失,形成NFTs。Kopke等[16]研究發現,在正常人腦中每個分子tau蛋白有2~3個分子的磷酸基團,但在AD患者的腦中,每個分子tau蛋白可有9~10個磷酸基團。因此,tau蛋白的異常過度磷酸化被認為是AD致病的關鍵因素。Ullrich(2010年)、Ghribi(2008年)、Schindowski(2008年)、Woodruff-Pak(2007 年)、Ghribi(2006年)等的研究均表明,高脂飲食能增加tau蛋白的表達和磷酸化。tau蛋白的磷酸化主要受蛋白激酶GSK-3β、MAPK等激酶的調控。Rahman研究表明高脂飲食能影響GSK-3β活性,因此GSK-3β激活可能是tau過度磷酸化的原因[17],并且Aβ的蓄積和tau蛋白的過度磷酸化密切相關,Aβ的蓄積能通過激活MAPK通路來增加tau蛋白的磷酸化。Ghribi等[18]用高脂飼料飼養7個月的兔后通過檢測PHF-1和AT8發現,高脂引發的tau過度磷酸化的位點在tau蛋白的Ser396或Ser404,而不是Ser202或Thr205,tau蛋白過度磷酸化同時伴隨著P-ERK的增加,而P-JNK與P-GSK-3β并沒有增加,提示高脂飲食引發的tau蛋白過度磷酸化可能是Aβ的蓄積激活了ERK而導致的,具體機制目前不明確,需進一步研究。
4 損傷神經可塑性
神經可塑性是神經系統的基本特征之一,包括突觸可塑性、神經發生、神經網絡重組和功能腦區轉移等,其中最突出的表現為突觸可塑性。突觸可塑性是指突觸在一定條件下通過改變形態而調整功能的能力,在神經發育、成熟及學習記憶等眾多的生理功能中起重要作用,可分為突觸結構和效能可塑性兩方面。前者指突觸形態的改變、新突觸連接形成和突觸功能的建立;后者是指突觸的反復活動引致突觸傳遞效率的增加或降低,包括長時程增強(LTP)和長時程抑制(LTD)[19]。
近年來大量的研究報道,高脂飲食可以改變突觸效能可塑性和突觸結構可塑性,從而損傷學習記憶。首先,高脂飲食能夠損傷突觸功能可塑性,Alexis[20]的研究證明高脂飼料飼養8個月后抑制大鼠海馬 CA1區的LTP。Gault(2010年)、Mattson(2010年)等也研究證明高脂飲食可以抑制LTP。LTP形成機制復雜,高脂飲食抑制LTP的機制尚不完全清楚,目前研究報道,高脂可抑制突觸前synapsin I磷酸化,抑制與LTP及學習記憶密切相關的核轉錄因子CREB的磷酸化,并且降低BDNF的表達[21],從而損傷突觸可塑性。高脂飲食也明顯損傷突觸結構可塑性。任姍姍等[22]的研究表明高脂飲食能導致正常小鼠突觸體數量衰減和突觸體膜流動性下降。Mattson(2010年)研究發現高脂飼養8個月后可以降低海馬突觸棘密度。Molteni報道,高脂飼養6個月能夠降低海馬中GAP43的mRNA和蛋白含量。Mattson[23]研究發現,高脂飲食能使大鼠海馬CA1區的微管相關蛋白(MAP2)染色減弱,MAP2的分布與海馬特別是CA1-CA3區神經元的完整性密切相關,它的減少表明海馬神經元受損。因此,高脂飲食能夠明顯損傷突觸可塑性。
5 損傷膽堿能神經系統
乙酰膽堿在神經系統中廣泛分布,對控制腦血流量、睡眠-覺醒循環以及調節認知能力和學習記憶過程都有重要作用。膽堿能系統的功能紊亂與進行性記憶缺失直接相關。前腦基底核的膽堿能細胞丟失是AD的典型特征,膽堿能神經遞質釋放的損傷能引起Aβ沉積,增加tau蛋白磷酸化,導致AD。Ullrich等的研究表明,給予高脂飲食5個月的大鼠與空白組相比,血漿膽固醇水平明顯上升、學習記憶能力受損,并且高膽固醇大鼠腦基底核膽堿能神經元的數目減少,皮層乙酰膽堿含量降低,從而損傷膽堿能神經元[24]。Gonzalo-Ruiz等[25]也報道高膽固醇飲食能加劇Aβ聚集造成的老年鼠大腦皮層膽堿能神經元數目的減少。因此,高脂可通過損傷膽堿能神經系統而導致AD發生、發展。
綜上所述,高脂飲食能通過增加Aβ產生,誘導神經炎癥,促進tau蛋白過度磷酸化,損傷突觸可塑性及膽堿能神經系統而降低認知功能,成為AD發病的危險因素。因此,合理的飲食可防止高脂引起認知障礙,減少AD發病幾率。
[1]鄭元元,孫 蘭.阿爾采末病非人靈長類動物模型[J].中國藥理學通報,2005,21(6):649-53.
[1]Zheng Y Y,Sun L.Progress in the primate models of Alzheimer’s disease[J].Chin Pharmacol Bull,2005,21(6):649-53.
[2]葉 偉,唐孝威.老年性癡呆癥發病機制及其防治措施的研究進展[J].國外醫學遺傳學分冊,2005,28(6):379-81.
[2]Ye W,Tang X W.Research progress on the pathogenesis and prevention measures of senile dementia[J].Section Genet Foreign Med Sci,2005,28(6):379-81.
[3]Zhang X C,Dong F,Jun R,et al.High dietary fat induces NADPH oxidase-associated oxidative stress and inflammation in rat cerebral cortex[J].Exp Neurol,2005,191:318-25.
[4]Anstey K J,Lipnicki D M,Low L F.Cholesterol as a risk factor for dementia and cognitive decline:a systematic review of prospective studies with meta-analysis[J].Geriatr Psychiatry,2008,16(5):343-54.
[5]Ullrich C,Pirchl M,Humpel C.Effects of cholesterol and its 24SOH and 25-OH oxysterols on choline acetyltransferase-positive neurons in brain slices[J].Pharmacology,2010,86(1):15-21.
[6]Jaya Prasanthi R P,Schommer E,Thomasson S,et al.Regulation of β-amyloid levels in the brain of cholesterol-fed rabbit,a model system for sporadic Alzheimer’s disease[J].Mech Ageing Dev,2008,129(11):649-55.
[7]Evin G,Barakat A,Masters C L.BACE:Therapeutic target and potential biomarker for Alzheimer’s disease[J].Int J Biochem Cell Biol,2010,42(12):1923-6.
[8]Marquer C,Devauges V,Cossec J C,et al.Local cholesterol increase triggers amyloid precursor protein-Bace1 clustering in lipid rafts and rapid endocytosis[J].FASEB J,2011,25(4):1295-305.
[9]Leclerc E,Sturchler E,Vetter S W.The S100B/RAGE axis in Alzheimer’s disease[J].Cardiovascul Psych Neurol,2010,2010:539-81.
[10]Waldron E,Jaeger S,Pietrzik C U.Functional role of the low-density lipoprotein receptor-related protein in Alzheimer’s disease[J].Neurodeg Dis,2006,3(4-5):233-8.
[11]Jeynes B,Provias J.Evidence for altered LRP/RAGE expression in Alzheimer lesion pathogenesis[J].Curr Alzheimer Res,2008,5(5):432-7.
[12]Carrasquillo M M,Belbin O,Zou F,et al.Concordant association of insulin degrading enzymegene(IDE)variants with IDE mRNA,Aβ,and Alzheimer’s disease[J].PLoS One,2010,5(1):e8764.
[13]Kurochkin I V,Goto S.Alzheimer’s β-amyloid peptide specifically interacts with and is degraded by insulin degrading enzyme[J].FEBS Lett,1994,345(1):33-7.
[14]郝麗娜,張慶柱,于天貴.非甾體類抗炎藥抗阿爾采末病作用的研究進展[J].中國藥理學通報,2008,24(8):988-92.
[14]Hao L N,Zhang Q Z,Yu T G.Research progress in the effects of nonsteroidal anti- inflammatory drug on Alzheimer’s disease[J].Chin Pharmacol Bull,2008,24(8):988-92.
[15]Granholm A C,Bimonte-Nelson H A,Moore A B,et al.Effects of a saturated fat and high cholesterol diet on memory and hippocampal morphology in the middle-aged rat[J].Alzheimer’s Dise,2008,14(2):133-45.
[16]Kopke E,Tung Y C,Shaikh S,et al.Microtubule-associated protein tau.Abnormal phosphorylatjon of a nonpaired helical filament pool in Alzheimer disease[J].Biol Chem,1993,268(32):24374- 84.
[17]Zhang T,Pan B S,Sun G C,et al.Diabetes synergistically exacerbates postsroke demetia and tau abnormality in brain[J].Neurochemistry International,2010,56(8):955-61.
[18]Ghribi O,Larsen B,Schrag M.High cholesterol content in neurons increases BACE1,β-amyloid,and phosphorylated tau levels in rabbit hippocampus[J].Experimental Neurology,2006,200(2):460-7.
[19]王 佩,王海祥.突觸可塑性與學習記憶[J].腦與神經疾病雜志,2008,16(5):651-3.
[19]Wang P,Wang H X.Synaptic plasticity and cogntion[J].J Brain Nervous Dis,2008,16(5):651-3.
[20]Stranahan A M,Norman E D,Lee K,et al.Diet-induced insulin resistance impairs hippocampal synaptic plasticity and cognition in middle-aged rats[J].Hippocampus,2008,18(11):1085-8.
[21]Moltrni R,Barnard R J,Ying Z,et al.A high-fat,refined sugar diet reduces hippocampal brain-derived neurotrophic factor,neuronal plasticity,and learning[J].Neuroscience,2002,112(4):803-14.
[22]任姍姍.高脂膳食對小鼠學習記憶能力和突觸可塑性的影響[J].中國老年學雜志,2010,13(30):1824-6.
[22]Ren S S.Effects of high fat diet on spatial learning and hippocampal synaptic plasticity[J].Chin J Gerontol,2010,13(30):1824-6.
[23]Mattson M P.The impact of dietary energy intake on cognitive aging[J].Front Aging Neurosci,2010(2):5.
[24]Celine U,Pirchl M,Humpel C.Hypercholesterolemia in rats impairs the cholinergic system and leads to memory deficits[J].Mol Cell Neurosci,2010,45(4-13):408-17.
[25]Gonzalo-Ruiz A,Sanz J M,Arévalo J,et al.Amyloid beta peptideinduced cholinergic fibres loss in the cerebr cortex of the rat is modified by diet high in lipids and by age[J].J Chem Neur,2004,29(2005):31-48.
