美國對高風險醫療器械的審批
2013-03-03 01:51:16常永亨
中國醫療器械雜志 2013年2期
關鍵詞:產品
常永亨
中國醫藥國際交流中心,北京市,100082
美國對高風險醫療器械的審批
【作 者】常永亨
中國醫藥國際交流中心,北京市,100082
在各國對醫療器械進行監督管理的實踐中,監管的嚴格程度和需要通過的法規程序是根據醫療器械的風險等級來確定的。該文簡要介紹了美國對高風險醫療器械進入市場的法規要求。
醫療器械;法規;風險;上市前審批
1 美國的醫療器械分類
美國對醫療器械進行監管的法律依據是《聯邦食品、藥品與化妝品法案》(Federal Food, Drug, and Cosmetic Act)中的醫療器械修正案(Medical Device Amendments)。根據該修正案,美國將醫療器械按照風險等級劃分為三個管理類別,其宗旨是,對各類器械產品施以監管措施的深度,應以保證該類產品的安全性和有效性所必須為原則。
三個管理類別的劃分和監管方式是:
第一類是“普通管理”產品,即危險性小或基本無危險性的產品,例如醫用手套、壓舌扳、手動手術器械、溫度計等。對這一類產品,企業在遞交FDA一2891表格后,產品即可上市。
第二類是“性能標準管理”產品,即具有一定危險性的產品,例如心電圖儀、超聲診斷儀、輸血輸液器具、呼吸機等。對這一類產品,在“普通管理”的基礎上增加實施依靠“性能標準”(performance standards)進行管理的措施,以保證產品的質量和安全性、有效性。
第三類是“用于支持或維持生命、對保護人類健康起至關重要的作用,或存在導致病痛或傷害的潛在、過度風險”的產品,例如人工心臟瓣膜、心臟起搏器、人工晶體、人工血管等。這是監管投入最深的醫療器械產品類別。對這一類產品,“普通管理”和“性能標準管理”措施都不足以保證就其“預設用途”(intended use)而言的安全性和有效性,因而所有屬于這一類別的醫療器械都要執行“上市前審批”(Premarket Approval — PMA)的要求。
根據這個劃分,美國市場上三個類別醫療器械的存在比例是:第一類約占全部醫療器械品種的25%,第二類約占全部醫療器械品種的55%,第三類約占全部醫療器械品種的20%。
2 上市前審批(PMA)
由食品藥品管理局(FDA)進行上市前審批是為保證第三類醫療器械的安全性和有效性而對其施行科學審評措施的法定程序。基于與第三類器械相關聯的風險水平,FDA斷定單獨依靠一般或特別的管理不足以保證第三類器械的安全性和有效性,這些器械上市需要依據《聯邦食品、藥品與化妝品法案》中的第515條提出PMA申請。但某些在1976年醫療器械修正案發布前即已進入市場的第三類醫療器械(Class III preamendment devices )可以履行針對這些第三類器械的510(k)程序(基于歷史驗證)。
PMA是FDA所要求的最嚴格的器械上市申請,這類器械必須得到FDA批準方可上市。FDA給予PMA批準是基于以下判斷:企業提交的PMA申請資料已經包含了足夠的有效科學證據,足以保證該器械就其預設用途而言的安全性和有效性。PMA是準予申請人(或持有者)銷售某種器械的私人許可證,但PMA的持有者可以授權他人使用其有關資料。PMA申請人通常是指為得到FDA批準而提交的PMA申請資料的所有權人或授權使用人。申請人可以是個人、合伙企業、公司法人、協會、科學或學術團體、政府機關或社會組織,或其它法人實體,常常是產品的發明者或開發者,并且最終成為制造者。FDA 法規規定的PMA審查決斷時間為180天,但實際時間通常更長。在作出一個批準或否定決斷之前,FDA的一個相應的顧問委員會可能會召開一個公開會議來進行審評,然后給FDA提供一個是否應批準該申請的委員會建議。FDA將批準與否的決定通知申請人后,會在網上發布一個公告。這個公告有兩個作用:(1) 公布FDA作出該決定所依據的資料;(2) 給利益相關者提供一個在30天內向FDA訴求重新考慮該決定的機會。
關于上市前審批的規定位于聯邦規章法典“Code of Federal Regulations”(CFR)第21部第814部分—“上市前審批”中。《聯邦食品、藥品與化妝品法案》第501(f)節規定,不符合PMA要求的第三類器械不得進入市場。
3 需要履行PMA的情形
PMA適用于醫療器械分類中最嚴格的第三類器械。搜索FDA的產品分類數據庫可以找到器械產品的類別,可以得到器械的名稱、類別和一個連接到聯邦規章法典(CFR)的鏈接。CFR提供器械類型名稱、識別碼和分類信息。對于1976年醫療器械修正案發布之前已進入市場的第三類器械,CFR中給出了一個規則號。對需要PMA的第三類器械,CFR指出該器械為第三類,并提出一個要求取得PMA的生效期限。如果CFR的規定中指出“無要求上市前批準的生效期限”,則應提交第三類器械的510(k)。由于需要履行PMA的器械常常牽涉到一些新的概念,許多都不是醫療器械修正案之前上市的相同類型產品,因而在CFR中沒有分類規定。對這種情形,產品類別數據庫只給出器械類型名稱和產品代碼。
如果不清楚某一未分類器械是否需要PMA,可以使用“三字母產品代碼”搜索PMA數據庫和510(k)數據庫(點擊產品類別數據庫網頁頂端的超文本鏈接可以找到這兩個數據庫)。如果有FDA通過的510(k),且新器械與任意已獲通過的器械實質性等同,則提交 510(k)即可。此外,有的新型器械在產品類別數據庫中可能找不到。如果是一個高風險器械(具有第三類器械的特征)且發現不與任何一個第一、第二或適用于510(k)的第三類器械實質性等同,則該器械在美國上市前必須得到PMA批準。
對用于血液循環系統的器械,FDA有特別的審評要求。FDA的“生物、評價、研究中心”(Center for Biologic, Evaluation, Research - CBER)具有血液、血液制品、細胞療法以及與其相關的某些醫療器械方面的完整的專業知識。為了利用這些專業知識,凡用于血液采集和處理程序以及用于細胞療法的醫療器械的上市申請或研究申請(上市前告知、上市前審批和臨床研究器械豁免),都由CBER進行審評。雖然這些產品由CBER審評,但仍適用醫療器械法規。由CBER進行審評的醫療器械目錄可以上互聯網查到。CDRH的上市前審批指南和由CBER進行審評的具體醫療器械指南都可以聯網或與FDA接洽獲得。
4 PMA申請
PMA申請資料是申請人向FDA提交的用以展示第三類器械的安全性和有效性的符合科學性和法規性要求的文件。PMA申請資料中有行政管理成分,但好的科學成分和科學撰文是獲得PMA批準的關鍵。如果PMA申請資料中缺少管理清單上列明的成分,FDA將拒絕此PMA申請并停止進行深度的科學和臨床數據審查。如果PMA申請資料中缺少建立在充分的科學推論基礎上有效的臨床信息和科學分析,將會延遲FDA的審查和批準。如果PMA申請資料不完整、不準確、不系統、缺少關鍵信息、粗劣編制,也可導致延遲批準或駁回。器械制造商在向FDA寄送PMA申請資料前應先進行內部質量控制審查,以保證申請資料具有充分的科學性,并且編制完美。
資料的技術部分應包含數據、信息,使FDA能據此判斷批準或不批準該申請。技術部分又通常分為非臨床的實驗室研究和臨床調查兩部分。前者應包含微生物學、毒理學、免疫學、生物相容性、壓力、磨損、保存期限等實驗室試驗或動物試驗數據。用于安全性評價的非臨床研究必須按照21CFR Part 58(針對非臨床實驗室研究的優良實驗室規范)進行。后者應包含研究方案、安全性和有效性數據、不良反應和并發癥、器械失靈和替換、患者信息、患者投訴、所有個體受試者的數據列表、統計分析結果,以及臨床調查中獲得的任何其它信息。
5 PMA補充/修正(PMA Supplements and Amendments)
PMA補充/修正是申請人向FDA提交對最初的PMA申請資料做出變更/修正的文件。雖然PMA補充適用于已獲批準的PMA,但在很多情況下,對于一個還未獲批準的PMA或PMA的補充文件也可以進行修正。
具體說來,PMA補充是申請人就一個已獲PMA批準的器械所發生的對安全性和有效性有影響的變化所補充提交的資料。而就正在審查中的PMA補充資料再次向FDA提交的補充資料叫做補充資料的修正。PMA修正包括對一個批準前的PMA或PMA補充所提交的所有補充資料,或就一個批準后的PMA或PMA補充所做的后續反應。這種情形與“臨床研究器械豁免”(Investigational Device Exemption - IDE) 情形的補充資料定義用語稍有不同:IDE補充是指一個IDE批準后對其提交的任何補充資料,IDE修正是指一個IDE獲得批準前對其提交的任何補充資料。
6 PMA審評程序
對PMA申請進行的審評包含四個步驟:
(1) 由FDA工作人員對申請資料的完整性進行形式審查和有限的技術審查(立檔審查);
(2) 由適任的FDA審查人員進行深入的科學審查、形式審查和質量體系審查;
(3) 由適任的專家小組進行審評并提出建議(專家組審評);
(4) 最終審議、歸檔、發出FDA的決定通知。
6.1 PMA申請的立檔(21CFR.814.42)
在進行形式審查和有限的技術審查過程中,FDA依據FD&C Act和PMA法規(21CFR814)規定的資料要求以及拒絕立檔政策對申請資料進行審查,以判斷一項PMA申請是否應予立檔。FDA制定了專門的資料檢查表用于PMA申請的立檔決定。
對一項申請予以立檔意味著FDA已作出門檻判斷——該申請資料的完整度已足以開始進行深入審評。FDA在收到PMA申請的45天內,將通知申請人是否已對該申請立檔。通知函中有PMA參考號和FDA對該PMA申請的立檔日期。如果適用于加快審評情形,也會在此時告知。立檔日期是表示審評機構收到一項接受立檔的PMA申請的日期,180天的PMA審評期限從這一天開始算起。
對于不符合最低門檻要求的申請,FDA不予立檔進行實質性審評。如果資料中的信息或數據表達含糊、不完整,或經不起嚴格的科學審評,FDA可以認為該PMA申請不完整,不予立檔。
對于不予立檔的PMA申請,FDA會將拒收的原因通知申請人。通知中會給出申請資料中導致不予立檔的缺陷,也會給出PMA參考號。FDA會建議制造商必須補充哪些資料,或采取哪些步驟,使申請得以符合立檔要求。對于不予立檔的申請,申請人可以:
(1) 補充符合FD&C Act第§515(c)(1)(A)-(G)節和21 CFR 814.20要求的資料后,重新提交PMA申請。重新提交的申請中必須標明原始提交的申請中的PMA參考號。如果重新提交的PMA申請被接受立檔,則FDA接受重新提交的申請的日期為立檔日期。
(2) 于收到拒絕立檔通知的10個工作日內,書面提出與器械審評辦公室主任就FDA不予立檔的決定進行一次非正式會談的請求。FDA將在收到請求后的10個工作日內進行該次非正式會談,并在非正式會談后的5個工作日內作出立檔與否的決定。如果FDA此時接受對該PMA申請立檔,則作出接受立檔決定的日期為立檔日期。如果FDA不改變拒絕立檔的決定,申請人可向CDRH主任提出再次審議請求。CDRH主任的決定將作為公正審評意義上的最終行政裁定。
6.2 深入審查
接受立檔以后,FDA開始對PMA申請進行實質性審查。審查過程中,FDA如果需要申請人補充任何為使FDA完成審查所必須的信息,會向申請人發出重要缺陷或輕微缺陷通知書。在PMA立檔后的100天內,申請人可以請求與FDA會談,討論對該申請的審查情況。從“PMA百日會談程序及后續缺陷指南——CDRH和企業使用,最終版”中可以找到“百日會談”的程序。如果申請人主動提交或應FDA的要求提交了PMA修正申請,其中有出自于先前未報告過的研究的重要新數據、出自于先前報告過的研究的重要更新數據、對先前提交的數據所做的詳細的新分析結果、或先前遺漏的重要的必須信息,審評期限可延至180天。
6.3 專家組審評
FDA可以委托一個外部專家組(顧問委員會)對PMA申請進行審評。一般情況下,同一種器械中第一個提交的PMA申請,FDA都會交給一個適任的顧問組進行審評和提出建議。此后,一旦FDA確信在判斷該種醫療器械的安全性和有效性方面所面臨的關健問題已經明了,并且FDA已具備了處理這些問題的能力,便不再把后面接受的同種器械的PMA申請交給專家組審評,除非某一個特別的申請中存在一個只有交給專家組審評才能得到最佳處理的問題。FDA可以將PMA申請資料或其中的有關部分交給顧問委員會中的每一個成員進行審評。審評過程中,FDA可能會與申請人或顧問委員會溝通,以便對顧問委員會成員可能提出的問題做出反應,或向顧問委員會提供補充信息。與申請人和顧問委員會進行的所有溝通,FDA都會存檔。
對交給顧問委員會進行審評的PMA項目,顧問委員會必須按照21 CFR 14的規定舉行一次公開的審評會。審評完成后,顧問委員會必須向FDA提交一份包括委員會對該PMA項目的建議和提出建議的根據的最終報告。顧問委員會報告和建議的形式可以是一份由委員會主席簽名的會議記錄。
FDA在綜合考慮會議記錄、專家組建議和其它信息之后對PMA申請做出最終決斷。FDA會把是否同意專家組建議以及還需申請人補充哪些信息的情況通知申請人(可予批準和不予批準的決定)。如果該申請可予批準,申請人必須同意“批準條件”。
6.4 批準通知
FDA應在PMA立檔日之后的180天內完成該項PMA審評以及顧問委員會報告和建議,并發出下列之一:
(1) 依據§814.44(d)的批準令;
(2) 依據§814.44(e)的可予批準信函;
(3) 依據§814.44(f)的不可批準信函;
(4) 依據§814.45的否決令。
6.4.1 批準令
FDA于審議了顧問委員會的最終報告后,如果沒有發現PMA否決條款(§814.45)中的任何適用項,即會向申請人頒發該PMA已獲批準的命令。FDA以產品說明(labeling)的最終文稿作為批準PMA申請的基礎,條件是申請人要在產品進入市場前向FDA提交一份最終印刷版的產品說明。FDA會將該PMA的批準情況向社會公布,把宣布批準決定和作為批準依據的安全性有效性資料概要(SSED)的公告放到互聯網上。概要中包括該器械可能存在的對健康的任何不良作用。公告還給申請人和其它利益相關者提供了一個依據FD&C Act第515(d)(3)節的規定對該項FDA批準決定提請行政復議的機會。每個季度,FDA還會在聯邦公報(Federal Register)上刊登一個本季度發布過的批準公告的目錄。依據§814.9,每一個批準公告發布后,該PMA檔案中的數據和資料即可供公眾查閱。
6.4.2 可予批準信函
如果一項PMA申請實質上符合FD&C Act的要求,并且FDA確信當申請人補交了有關具體資料或同意了具體條件以后可以批準該申請,FDA會向申請人寄發一封可予批準信函。信函中會說明作為批準的前提要求申請人補交的資料或要求申請人滿足的條件。例如,FDA可以提出下列批準條件:
(1) 提交批準信函中列出的一些資料,比如產品說明終稿;
(2) 由FDA對生產場所進行一次現場審查,證明生產設施、方法和質量控制符合質量體系規定(21 CFR 820),并且,適用時,查證與批準該PMA相關的一些記錄;
(3) 依據FD&C Act第515(d)(1)(B)(ii)節或520(e)節,對該器械的銷售、流通或使用設置的一些限制。批準后可以要求申請人同意批準后繼續進行研究,僅限于處方使用,或要求申請人對器械的使用者進行培訓。還可能要求申請人進行上市后監督、跟蹤。
申請人回復可予批準信函時,可以:
(1) 按要求做出修正;
(2) 將可予批準信函視為對該PMA申請的否決(21 CFR 814.45),并以發再議請求書(21 CFR 10.33)的方式要求行政復議[FD&C Act第515(d) (3)節];
(3) 撤回該PMA申請。
6.4.3 不可批準信函
如果FDA確信由于不符合§814.45(a)中的一條或幾條因而不能批準該申請,或由于申請資料中缺乏關鍵信息而無法做出批準決定時,FDA會向申請人寄發一封不可批準信函。不可批準信函會說明申請資料中存在的缺陷,包括依據FD&C Act第515(d)(2)(A)-(E)節的規定而否決批準的每一個理由。可行的情況下,FDA會告知使該PMA申請變為可予批準的必要做法。
申請人回復不可批準信函時,可以:
(1) 按要求做出修正[依據§814.37(c)(1),此類修正屬于重要修正];
(2) 將不可批準信函視為對該PMA申請的否決(21 CFR 814.45),并以發再議請求書(21 CFR 10.33)的方式要求行政復議[FD&C Act第515(d) (3)節];
(3) 撤回該PMA申請。
在下列情況下,FDA將PMA申請視為申請人主動撤回:
(1) 在FDA寄發修正要求后180天內,申請人未對該要求做出書面答復;
(2) 在FDA寄發可予批準或不可批準信函后180天內,申請人未對信函做出書面答復;
(3) 申請人向FDA提交撤回該PMA申請的書面通知。
6.4.4 否決令
如果申請人不遵守PMA法規的要求,或者FDA認為FD&C Act第515(d)(2)(A)-(E)節中關于否決PMA的任何一條理由適用,FDA可以否決該PMA申請。此外,FDA還可以依下列任一理由否決PMA:
(1) 在重要事實上存在不實命題;
(2) 器械的說明文字不符合第801部分關于說明的規定,或第809部分關于人用體外診斷產品的規定;
(3) 申請人不允許FDA審查人員到器械生產現場審查生產設施和管理控制措施,或獲取、復制與該申請獲得批準相關的全部現場記錄;
(4) PMA資料中敘述的一個重要的非臨床的實驗室研究情況不符合21 CFR 58中的GLP規定,并且沒有解釋該項不符合情況的原因。或者解釋了原因,但該實驗所用的方法與GLP法規要求之間的差別無法支持該研究結果的有效性;
(5) PMA資料中敘述的任一人體臨床研究項目,未能按照21 CFR 56關于機構審查委員會的規定或21 CFR 50關于知情同意的規定執行,因而受試人的權益或安全沒有得到充分保護。
FDA依據§814.17發出對PMA申請的否決令。否決令中會告知申請人資料中存在的缺陷,包括依據FD&C Act第515(d)(2)節和第814部分的規定而予以否決的每一個理由。可行的情況下,也會告知申請人以可予批準的形式提交該PMA申請的必要做法。否決令中還包含申請人可以依據FD&C Act第515(d)(3)節的規定要求復議的告知。
7 PMA收費
自2002年10月1日起,FDA開始收取PMA審查費。2002年10月26日,醫療器械審查收取用戶費(user fee)的規定和2002現代化案(Modernization Act of 2002)被寫入法律。這項法律批準FDA就醫療器械審查收取費用(包括PMA審查)。
2012財政年度(2011年10月1日至2012年9月30日),對PMA申請的收費規定如表1所示。
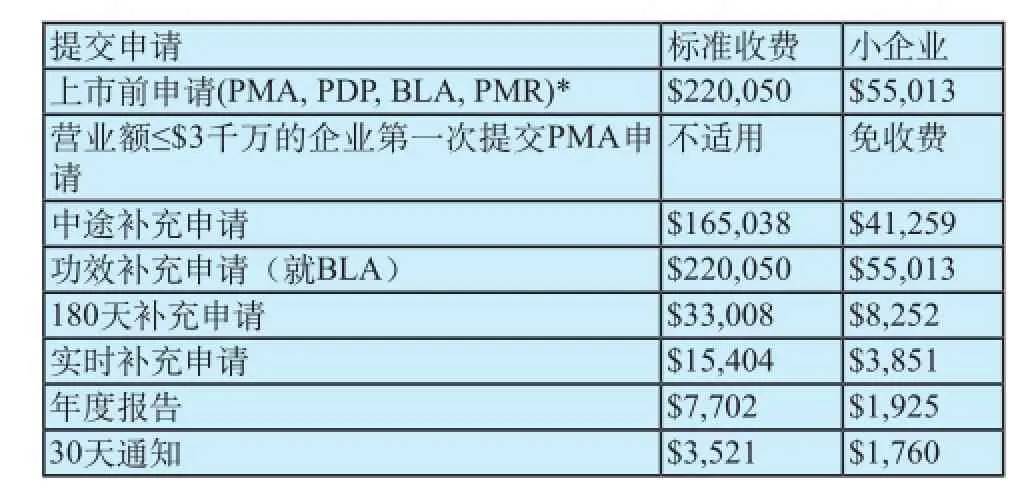
表1 2012財年器械審評用戶費(美元)Tab.1 FY 2012 device review user fees($)
表1中所列的申請項目都必須按規定交費,除非申請人有資格免除費用或得到豁免。小企業有資格減少費額。必須在提交申請的同時或之前交費。如果沒有付清所有欠費,FDA將按申請資料不完整處理,不予立檔。
根據不同國家的具體情況,各國對高風險醫療器械的審批方式和法規要求不盡相同。以上美國對高風險醫療器械審批的做法可供我國醫療器械監管中參考或借鑒,也可供欲向美國出口醫療器械的企業參考。
USA Approval of High-risk Medical Device
【 Writer 】Chang Yongheng
China center for pharmaceutical International excharge, Beijing, 100082
During the practice of supervision and management of medical device in different countrie, the strict regulatory and needs of the program by the regulations is to be determined by the risk level of the medical equipment.This paper brie fl y describes the regulatory requirements of the United States to enter the market for high-risk medical device.
medical device, act, risk, pre-market approval
TH77
10.3969/j.issn.1671-7104.2013.02.016
A
1671-7104(2013)02-0132-05
2013-01-13
常永亨,主任,E-mail: cyh0010532@sina.com
猜你喜歡
現代裝飾(2022年4期)2022-08-31 01:39:32
現代裝飾(2022年3期)2022-07-05 05:55:06
物流技術與應用(2022年5期)2022-06-17 06:01:38
快樂語文(2021年36期)2022-01-18 05:48:46
金橋(2021年4期)2021-05-21 08:19:22
中國化妝品(2018年6期)2018-07-09 03:12:40
中國化妝品(2018年6期)2018-07-09 03:12:32
Coco薇(2015年1期)2015-08-13 02:23:50
汽車維修與保養(2015年6期)2015-04-17 03:31:50
玩具(2009年10期)2009-11-04 02:33:14
