非諾貝特對壓力超負荷中期大鼠心肌能量代謝和心室重構的影響*
2013-04-19 06:38:45何燕鄔江濤鐘國強吳龍飛李醒三王炎陳卓宏閉奇
中國循環雜志 2013年2期
關鍵詞:手術
何燕,鄔江濤,鐘國強,吳龍飛,李醒三,王炎,陳卓宏,閉奇
·基礎與實驗研究·
非諾貝特對壓力超負荷中期大鼠心肌能量代謝和心室重構的影響*
何燕,鄔江濤,鐘國強,吳龍飛,李醒三,王炎,陳卓宏,閉奇
目的:研究非諾貝特對壓力超負荷中期大鼠心肌能量代謝和心室重構的影響。
非諾貝特;肥厚;重構;能量代謝;壓力超負荷
(Chinese Circulation Journal, 2013,28:140.)
心臟壓力超負荷可導致一系列復雜的級聯信號瀑布激活,產生適應性應答以維持正常心臟輸出。這種適應包括心肌細胞質量、基因表達和代謝的調整。在結構水平,表現為心臟肥大;在轉錄水平,表現為胚胎期基因再表達,而多種成人期基因表達下調[1];在代謝水平,表現為心肌能量底物由以脂肪酸β氧化為主轉變為以葡萄糖酵解為主,這與過氧化物酶體增殖物 激 活 受體 α(peroxisome proliferator activated receptors α, PPARα)、 中 鏈 脂 酰 輔 酶 A 脫 氫 酶 (medium-chain acyl-CoA dehydrogenase, MCAD)、 肌型 肉堿棕櫚 酰轉移酶 -1(muscle carnitine palmitoyl transferase-1, MCPT-1) 活性降低密切相關[2,3]。研究發現 PPARα 激活劑在壓力超負荷動物模型早期短暫應用即可減輕心肌肥厚[4],但另有研究發現后期長期應用反而加重心肌肥厚[5]。本研究將觀察 PPARα激活劑非諾貝特對壓力超負荷中期大鼠心肌重構的影響,有助于探索其在心肌肥厚不同病理生理階段的作用機制。
1 材料和方法
實驗動物:2010-07 至 2011-03,選擇 8 周齡健康雄性 Wistar大鼠 42 只(廣西醫科大學動物實驗中心提供),體重 200~250 g。
分組、建立大鼠壓力超負荷模型及評價 : 8 周齡健康 Wistar大鼠,參照文獻 [6]采用腹主動脈縮窄術建立壓力超負荷模型后,隨機分為2組:對照組(14只)和非諾貝特組(14只);另采用假手術(在腹主動脈相同部位穿線但不結扎)建立假手術組(14 只)。術后抗感染4天,觀察4周后,各組分別取6只大鼠,麻醉固定后,經右頸總動脈插入心導管,連接生理信號采集處理儀,記錄左心室收縮壓、左心室舒張末壓、左心室壓力上升和下降最大速率,評價模型建立情況。
壓力超負荷大鼠模型的干預 : 模型成功建立后,非諾貝特組予非諾貝特 150 mg/(kg·d)溶于 2 ml生理鹽水后灌胃喂食;對照組、假手術組予等容積生理鹽水灌胃喂食。干預8周。
留取血清和心肌組織標本 : 干預結束后,各組大鼠留取血清;開胸取出心臟,預冷生理鹽水沖洗,濾紙吸干水份后稱取左心室、右心室濕重,計算左心室濕重 /體重、右心室濕重 /體重,分別為左心室和右心室心肌質量指數。心底部心肌組織分裝于4%甲醛液中固定,心尖部分于液氮中速凍后存于 -80℃超低溫冰箱,待分子生物學實驗。
心肌組織病理學形態 : 心肌組織按常規制作病理切片,做馬松(Masson)染色,光鏡觀察細胞形態和膠原纖維分布,采用 HP IAS2000 計算機圖像分析系統分析并計算膠原容積分數。
觀察心肌超微結構及心肌線粒體損傷程度分級半定量分析 : 病理切片經醋酸雙氧鈾、枸櫞酸鉛雙重染色,電鏡觀察心肌超微結構尤其是線粒體的變化,并照片分析,按照 Flameng 評分[7]進行損傷程度分級。
血清和心肌游離脂肪酸測定 : 按 Nixon 等[8]的方法,根據吸光率計算血清和心肌勻漿中游離脂肪酸的水平。
免疫印跡檢測PPARα、MCAD、MCPT-1蛋白表達。
半定量逆轉錄聚合酶鏈反應(RT-PCR)檢測PPARα、MCAD、MCPT-1 基因 表達 : 設 計并由 上海生 物 工程公 司合成 PPARα、MCAD、MCPT-1引 物,Trizol法 提 取 左 心 室 心 肌 組 織 總 核 糖 核 酸(RNA),取 2 μg RNA 進行逆轉錄反應合成脫氧核糖核酸(cDNA),進行 RT-PCR 后,取 PCR 產物 4 μl進行凝膠電泳并經成像系統分析,計算 PPARα、MCAD、MCPT-1 與甘油醛 -3- 磷酸脫氫酶 (GAPDH)信使核糖核酸 ( mRNA)的灰度比值。
統計學方法: 各組數據均采用 SPSS 13.0 統計軟件分析,計量資料采用均數 ± 標準差(x±s)表示,多組間比較采用方差分析,進一步兩兩比較采用獨立t檢驗,以P<0.05 為差異有統計學意義。
2 結果
2.1 實驗大鼠一般情況
研究期間,假手術組死亡 2 只,對照組死亡 3 只,非諾貝特組死亡3只:死因多為感染、出血、呼吸衰竭和心力衰竭。最終,假手術組(12 只)、對照組(11 只)、非諾貝特組(11 只)到達研究終點。
2.2 壓力超負荷大鼠模型建立的評價
腹主動脈縮窄術后4周測定血流動力學指標,對照組、非諾貝特組左心室收縮壓、左心室舒張末壓較假手術組增高(P<0.05),左心室壓力上升和下降最大速率無明顯變化(P>0.05);對照組與非諾貝特組間比較差異無統計學意義(P>0.05,表1)。初步判斷壓力超負荷中期大鼠模型成功建立。
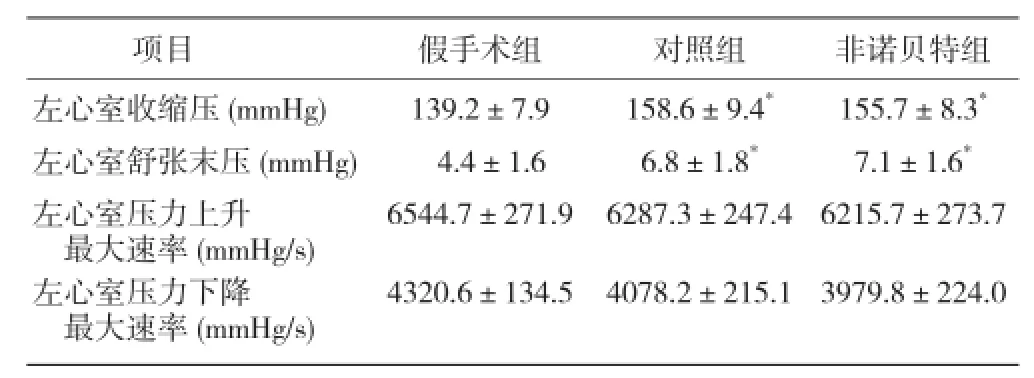
表1 3 組腹主動脈縮窄術后 4 周血流動力學指標(x±s, n=6)
2.3 3 組心肌重構指數的變化
對照組及非諾貝特組左心室質量指數較假手術組明顯增高;非諾貝特組與對照組比增高程度低于對照組(P均 <0.05),差異均有統計學意義;3 組間右心室質量指數差異均無統計學意義(P>0.05,表2)。
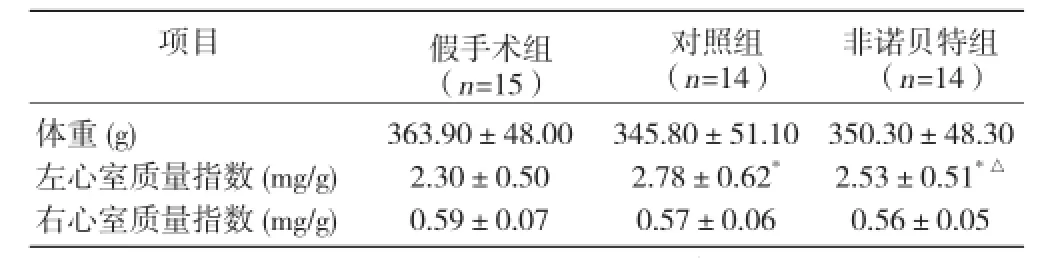
表2 3 組干預后心肌重構指數變化(x±s)
2.4 3 組心肌形態及膠原纖維分布
心肌組織馬松染色顯示,對照組、非諾貝特組心肌粗大、間質膠原纖維增生,進一步計算膠原容積 分 數, 對 照 組、 非 諾 貝 特 組 分 別 為 3.14±0.37、2.47±0.55,較假手術組 1.78±0.42 均增高(P<0.05),但非諾貝特組較對照組降低(P<0.05),差異均有統計學意義。圖1

圖1 大鼠心肌的組織病理學比較。 A: 假手術組心肌細胞正常,排列整齊,無間質膠原纖維增生;B: 對照組心肌稍顯粗大,排列尚整齊,間質膠原纖維略顯增生;C: 非諾貝特組心肌纖維接近正常,排列整齊,間質膠原纖維增生程度輕微。馬松染色
2.5 3 組心肌超微結構及線粒體損傷程度分級半定量分析
電鏡觀察心肌超微結構,對照組、非諾貝特組心肌細胞及線粒體腫脹、破壞,非諾貝特組輕于對照組(圖2);作線粒體損傷程度 Flameng 評分,對照組、非諾貝特組心肌線粒體損傷程度分別 為 1.7±0.5、1.0±0.4,均高 于假 手術 組 0.4±0.3(P<0.05),非諾貝特組則低于對照組(P<0.05),差異均有統計學意義。
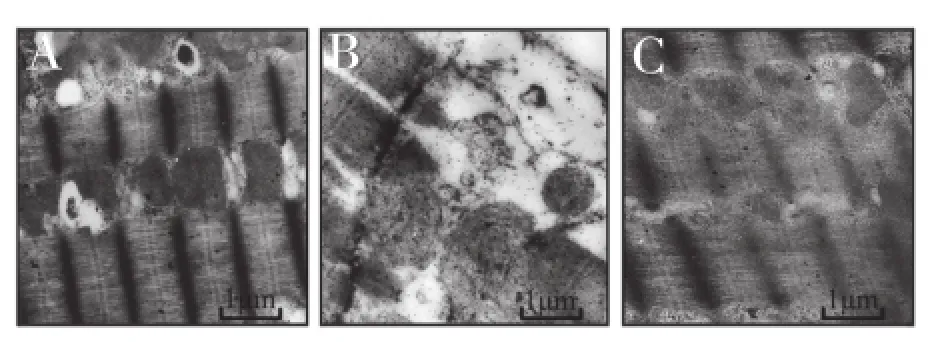
圖2 大鼠心肌的超微結構圖。A: 假手術組心肌細胞無腫脹,胞膜完整,肌絲整齊,肌小節明、暗帶清晰,線粒體整齊,膜完整,無腫脹,嵴突規則致密,線粒體間可見黑色糖原顆粒;B: 對照組心肌細胞腫脹,間有肌節溶解,伴肌膜破損,肌絲紊亂,線粒體明顯腫脹,基質透明,嵴斷裂或消失;C: 非諾貝特組心肌細胞輕度腫脹,肌絲基本整齊,肌節基本完整,線粒體輕度腫脹,基質基本完整,基質顆粒部分消失,嵴較規則,少部分斷裂
2.6 3 組血清及心肌游離脂肪酸的變化
對 照 組 血 清 游 離 脂 肪 酸(440.7±77.3)μmol/L高 于 假 手 術 組(325.8±62.5)μmol/L 和 非 諾 貝 特組(256.6±48.8)μmol/L;對照組心肌游離脂肪酸(336.0±42.5)μmol/L 高 于 假 手 術 組(187.5±44.7)μmol/L 和 非 諾 貝 特 組(192.4±46.5)μmol/L(P 均<0.05),差異均有統計學意義。
2.7 3 組心肌 PPARα、MCAD、MCPT-1 蛋白表達的變化
對照組、非諾貝特組心肌PPARα、MCAD、MCPT-1蛋白表達較假手術組均明顯下調;非諾貝特組較對照組均顯著上調(P均 <0.05),差異均有統計學意義。圖3
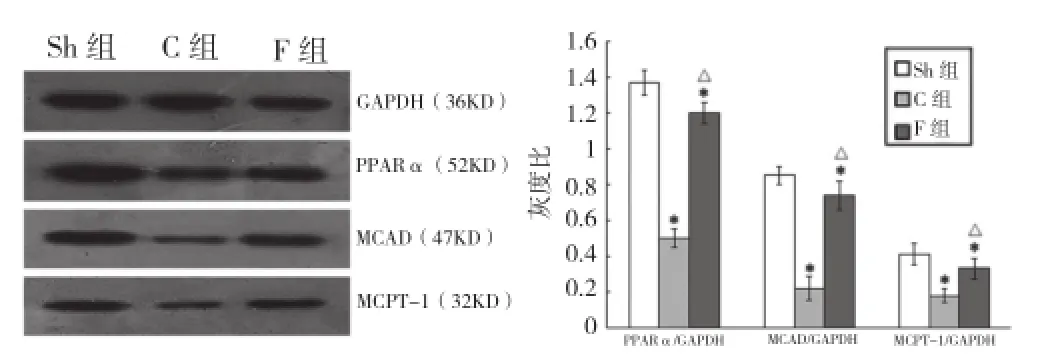
圖3 蛋白質印跡法檢測各組大鼠心肌 PPARα、MCPT-1、MCAD 蛋白表達。與假手術組比較*P<0.05;與對照組比較△P<0.05。GAPDH:甘油醛 -3-磷酸脫氫酶 PPARα:過氧化物酶體增殖物激活受體 α MCAD:中鏈脂酰輔酶 A 脫氫酶 MCPT-1:肌型肉堿棕櫚酰轉移酶 -1。Sh:假手術組;C:對照組;F:非諾貝特組
2.8 3 組 心 肌 PPARα、MCAD、MCPT-1 mRNA 基因表達的變化
對照組、非 諾 貝特組 心 肌 PPARα、MCAD、MCPT-1 mRNA 基因 表達 均較 假手術組明 顯下 調(P<0.05);非諾貝特組均較對照組顯著上調(P 均<0.05),差異均有統計學意義。圖4
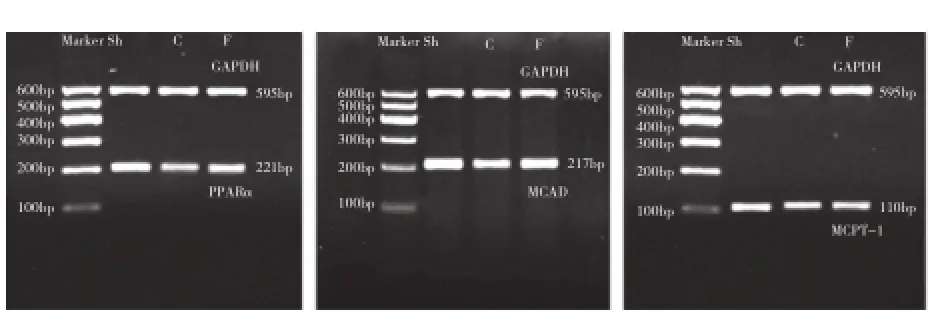
圖4 逆轉錄聚合酶鏈反應檢測各組大鼠心肌 PPARα、MCAD、MCPT-1 mRNA表達。余注見圖3
3 討論
壓力超負荷下,心肌在結構水平、代謝水平、基因表達水平均產生適應性調節,以維持正常的心臟功能[9-11]。腹主動脈縮窄誘導的壓力超負荷大鼠,心肌肥厚多始于術后 4 周[12],而心肌代謝模式轉換則始于術后 8 周[5]。基于現有的研究,PPARα 激活劑在壓力超負荷早期(2~4 周)短暫干預減輕心肌肥厚,后期(8~12 周 )長期干預可能加重心肌肥厚[5,12]。本研究 旨在觀察 PPARα 激活劑非 諾貝特在壓力超負荷中期對大鼠心肌肥厚的影響。選擇術后4周開始干預,持續到術后12周,覆蓋心肌重構和代謝重構發生發展的關鍵時期,預期研究結果相對具有代表性。本研究發現,術后4周,模型組血流動力學指標明顯變化,提示進入壓力超負荷中期;術后 12周,壓力后負荷的增加,左心室心肌為了維持正常的心臟輸出,發生了心肌肥厚;馬松染色證實,心肌正經歷由代償性肥厚向失代償性肥大和纖維化演變的進程。采用 PPARα 激活劑非諾貝特干預,可延緩壓力超負荷中期大鼠心肌肥厚的演變進程。
壓力超負荷下心肌代謝已經發生“能量缺乏”, 而心肌結構則逐漸出現“失調”,表現為心肌脂肪酸代謝水平和相關基因表達已下調,出現心肌肥厚和輕度纖維化,但光鏡下心肌結構完整,排列整齊[13]。為了避免持續的代謝“能量缺乏”進一步導致結構“失調”,提高心肌能量代謝水平尤為重要。本研究證實,模型組血清和心肌游離脂肪酸增高,可能是心肌脂肪酸β氧化轉變為以葡萄糖酵解為主的代謝模式,脂肪酸攝取減少,氧化能力減弱,導致脂肪酸蓄積,并產生自由基損傷線粒體、呼吸鏈和亞細胞器膜結構相關酶的功能,抑制線粒體、肌漿網和胞質酶體系中 Ca2+- 三磷酸腺苷(ATP)酶和 Na+-K+-ATP 酶等的活性[14,15],最終將抑制糖酵解模式的效率,導致心肌細胞和線粒體損傷逐漸加重,心肌能量進一步匱乏。
近年研究發現,PPARα可調控其下游編碼基因MCAD、MCPT-1。MCPT-1 是脂肪酸轉運到線粒體的關鍵酶,MCAD則是線粒體脂肪酸氧化第一步的關鍵酶。PPARα 激活劑可通過上調 PPARα 及其所調控的MCAD、MCPT-1基因表達,增強脂肪酸氧化,提高心肌能量代謝水平[16,17]。本研究中,心肌 PPARα、MCAD、MCPT-1蛋白和基因的表達在對照組均下調,在非諾貝特組則上調,提示非諾貝特可能通過上調脂肪酸氧化相關酶表達,改善代謝水平來減輕心肌肥厚。但另有研究表明長期激活可使脂肪酸過度氧化,增加心肌耗氧量,降低氧化效率,加劇心肌能量匱乏,加重心室重構。提示在機體能量代謝演變的不同階段,脂肪酸氧化與葡萄糖酵解模式之間的動態轉換,可能存在復雜的調控機制。總之,在壓力超負荷中期,PPARα 激活劑非諾貝特能夠改善心肌能量代謝,延緩心室重構,其機制可能與上調心肌 PPARα、MCAD、MCPT-1 蛋白和基因的表達增加脂肪酸氧化有關。
[1]Melenovsky V, Benes J, Skaroupkova P, et al. Metabolic characterization of volume overload heart failure due to aorto-caval fistula in rats. Mol Cell Biochem, 2011,354:83-96.
[2]Schwarzer M, Faerber G, Rueckauer T, et al. The metabolic modulators, Etomoxirand NVP-LAB121, fail to reverse pressure overload induced heart failure in vivo. Basic Res Cardiol, 2009,104:547-557.
[3]Rimbaud S, Ruiz M, Piquereau J, et al. Resveratrol improves survival, hemodynamics and energetics in a rat model of hypertension leading to heart failure. PLoS One, 2011,6:26391.
[4]Rose M, Balakumar P, Singh M. Ameliorative effect of combinationof fenofibrate and rosiglitazone in pressure overload-induced cardiac hypertrophy in rats. Pharmacology, 2007,80:177-184.
[5]Purushothaman S, Sathik MM, Nair RR. Reactivation of peroxisome proliferator-activated receptor alpha in spontaneously hypertensive rat: age-associated paradoxical effect on the heart. J Cardiovasc Pharmacol, 2011,58:254-262.
[6]羅 羽 慧 ,李 法 琦 ,梅 霞 ,等 .福 辛 普 利 對 實 驗 性 心 力 衰 竭 大 鼠心肌細胞凋亡和 Fas、Fas 配體基因表達的影響 .中國循環雜志 ,2004,19:66-70.
[7]Flameng W, Borgers M, Daenen W, et al. Ultrastructural and cytochemical correlates of myocardial protection by cardiac hypothermia in man. J Thorac Cardiovasc Surg, 1980,79:413-424.
[8]Nixon M, Chan SH. A simple and sensitive colorimetric method for the determinationof long-chain free fatty acids in subcellular organelles. Anal Biochem, 1979,97:403-409.
[9]Lionetti V, Stanley WC, Recchia FA. Modulating fatty acid oxidation in heart failure. Cardiovasc Res, 2011,90:202-209.
[10]Chaanine AH, Hajjar RJ. AKT signalling in the failing heart. Eur J Heart Fail, 2011,13:825-829.
[11]Lombardi R, Betocchi S. Aetiology and pathogenesis of hypertrophic cardiomyopathy. Acta Paediatr Suppl, 2002,91:10-14.
[12]Schultz Jel J, Glascock BJ, Witt SA, et al. Accelerated onset of heart failure in mice during pressure overload with chronically decreased SERCA2 calcium pump activity. Am J Physiol Heart Circ Physiol, 2004,286:1146-1153.
[13]韓瑜 ,韓變 梅 ,柳 明沫 ,等 . 心 肌膠 原及 代 謝 . 中 國循 環雜 志 , 2006,21:315-317.
[14]Purushothaman S, Renuka Nair R, Harikrishnan VS, et al. Temporal relation of cardiachypertrophy, oxidative stress, and fatty acidmetabolism in spontaneously hypertensive rat. Mol Cell Biochem, 2011,351:59-64.
[15]Jaswal JS, Keung W, Wang W, et al. Targeting fatty acid and carbohydrate oxidation--anovel therapeutic intervention in the ischemic and failing heart. Biochim Biophys Acta, 2011,1813:1333-1350.
[16]Balakumar P, Rose M, Singh M. Effect of fenofibrate in pressure overload-induced experimental cardiac hypertrophy. International Journal of Biological Chemistry, 2007,1:104-110.
[17]Lebrasseur NK, Duhaney TA, De Silva DS, et al. Effects of fenofibrate on cardiac remodeling in aldosterone-induced hypertension. Hypertension, 2007,50:489-496.
Effect of Fenofibrate on Myocardial Energy Metabolism and Ventricular Remodeling in Metaphase Pressure Overloaded Rats
HE Yan, WU Jiang-tao, ZHONG Guo-qiang, WU Long-fei, LI Xing-san, WANG Yan, CHEN Zhuo-hong, Bi Qi.
Department of Cardiology, The First Affiliated Hospital of Guangxi Medical University,
Nanning (530021), Guangxi, China
Objective: To investigate the effect of fenofibrate on myocardial energy metabolism and ventricular remodeling in metaphase pressure overloaded rats.Methods: The metaphase pressure overloaded animal model was established by aortic constriction in wistar rats and then randomly divided them into 3 groups, Control group and Sham operation (Sham) groupand the rats received normal saline in both groups, and Medication group, the rats received fenofibrate 150mg/(kg·d). n=14 in each group, all animals were treated for 8 weeks. The rats’ myocardial structure was observed by light microscope and the collagen volume fraction was calculated. The mitochondrial structure was studied by electron microscope and the Flameng score for myocardial mitochondrial injury was calculated. Serum and myocardial free fatty acid levels were examined, PPARα, MCAD and MCPT-1 gene and protein expressions were assessed by RT-PCR and western blot analysis respectively.Result: ① Compared with Sham group, both Control and Medication groups presented increased left ventricular weight index and collagen volume fraction; Medication group was much lower than that in Control group, P<0.05. ② Compared with Sham group, both Control and Medication groups showed increased Flameng score; Medication group was lower than that in Control group, P<0.05. ③ The serum and cardiac free fatty acid level in Medication group was lower than Control group, P<0.05. ④ The expression of PPARα, MCPT-1 and MCAD in both Control and Medication groups were down-regulated than that in Sham group; they were up-regulated in Medication group than that in Control group, P<0.05.Conclusion: Fenofibrate could increase fatty acid oxidation through up-regulating the expression of PPARα, MCPT-1 and MCAD, and therefore, reducing the serum and cardiac free fatty acid levels, extenuate mitochondrial injury, those are helpful to protect the cardiac function in metaphase of pressure overload rats.
Fenofibrate; Hypertrophy; Remodeling; Energy metabolism; Pressure overload
HE Yan, E-mail:hyxjwxy@126.com
2012-05-19)
(編輯:漆利萍)
*
廣西壯族自治區科技廳青年基金項目資助(桂科青 0728064),本研究受“廣西大型儀器協作共用網資助 ” 項目編號:635-2008-048
530021 廣西壯族自治區南寧市,廣西醫科大學第一附屬醫院 老年心內科
何燕 主任醫師 博士 主要研究方向為慢性心力衰竭機制及臨床 Email:hyxjwxy@126.com 通訊作者:何燕
R541
A
1000-3614(2013)02-0140-04
10.3969/j.issn.1000-3614.2013.02.018
方法 :采用腹主動脈縮窄術建立 Wistar 大鼠壓力超負荷模型,隨機分為對照組(14 只 )和非諾貝特組(14 只);另建立假手術組(14 只 )。假手術組和對照組給予生理鹽水、非諾貝特組給予非諾貝特 150 mg/(kg·d),干預 8 周后,留取大鼠心肌,光鏡觀察心肌結構,計算膠原容積分數;電鏡觀察線粒體結構,進行 Flameng 評分;測定血清及心肌游離脂肪酸水平;蛋白質印跡法、逆轉錄聚合酶鏈反應檢測過氧化物酶體增殖物激活受體 α(PPARα)、中鏈脂酰輔酶 A 脫氫酶 (MCAD)、肌型肉堿棕櫚酰轉移酶 -1(MCPT-1)蛋白及基因表達。
結果:①干預后,對照組、非諾貝特組心肌左心室質量指數、膠原容積分數較假手術組增高,非諾貝特 組則較對照組明顯降低(P 均 <0.05);②干預后,對照組、非諾貝特組心肌線粒體損傷 Flameng 評分高于假手術組,非諾貝特組評分低于對照組(P 均 <0.05);③干預后,非諾貝特組血清及心肌游離脂肪酸較對照組明顯降低(P均 <0.05);④干預后對照組、非諾貝特組心肌 PPARα、MCPT-1、MCAD 蛋白及基因表達均較假手術組下調, 非諾貝特組較對照組上調(P均 <0.05),差異均有統計學意義。
結論 :在壓力超負荷中期,非諾貝特通過上調大鼠心肌 PPARα、MCPT-1、MCAD 基因表達,增強脂肪酸氧化,而降低血清和心肌游離脂肪酸,減輕線粒體損傷,減輕左心室肥厚和心肌纖維化,有助于保護壓力超負荷下大鼠心臟功能。
猜你喜歡
環球時報(2022-12-23)2022-12-23 09:28:37
昆明醫科大學學報(2022年1期)2022-02-28 07:45:04
中老年保健(2021年11期)2021-08-22 03:13:36
昆明醫科大學學報(2021年2期)2021-03-29 07:42:46
河北畫報(2020年10期)2020-11-26 07:20:50
小學閱讀指南·低年級版(2017年1期)2017-03-13 20:07:35
中國衛生標準管理(2015年3期)2016-01-14 03:41:47
中國醫療美容(2015年1期)2015-07-12 10:06:38
鄭州大學學報(醫學版)(2015年2期)2015-02-27 14:50:54
西南軍醫(2014年5期)2014-04-25 07:42:48
