復方刺山柑膠囊的質量標準研究
2013-09-17 05:42:24楊偉俊羅玉琴薛文采
中國醫藥導報 2013年25期
袁 濤 楊偉俊 羅玉琴 薛文采 程 波
1.新疆維吾爾自治區克拉瑪依市中心醫院,新疆克拉瑪依 834000;2.新疆維吾爾自治區藥物研究所新疆維吾爾藥重點實驗室,新疆烏魯木齊 830004;3.中國科學院新疆理化技術研究所 中國科學院干旱區植物資源化學重點實驗室,新疆烏魯木齊 830011
復方刺山柑膠囊是通過臨床經驗結合現代藥物配伍篩選技術自主研發的復方藥物,處方由刺山柑、川西獐牙菜、波棱瓜子、大黃、苦荬菜、木香、兔耳草、唐古特烏頭、角茴香、甘草、金錢草、訶子、黃芪等多味藥材組成,具有清熱解毒、疏肝利膽、利濕退黃之功能,用于急性黃疸型肝炎、急性膽囊炎屬肝膽溫熱證者。課題組前期根據處方各藥味所含化學成分的理化性質結合本方功效,優化了復方刺山柑膠囊的提取工藝[1],并制備成膠囊劑。本文此其基礎上進行制劑的質量研究,為臨床用藥安全和有效提供質量控制標準。
1 材料與儀器
1.1 試驗材料
復方刺山柑膠囊(批號:100402、100403、100404、100405、100415、100416、100417、100723、100724);大黃素(中國藥品生物制品檢定所提供,批號:0756-200110);大黃酚對照品(中國藥品生物制品檢定所提供,含量測定用,批號:0796-200208);齊墩果酸對照品 (中國藥品生物制品檢定所提供,含量測定用,批號:110709-201206);黃芪甲苷對照品(中國藥品生物制品檢定所提供,含量測定用,批號:110781-200613);沒食子酸對照品(中國藥品生物制品檢定所提供,含量測定用,批號:110831-200803);木香對照藥材(中國藥品生物制品檢定所提供,批號:120921-201008);訶子對照藥材 (中國藥品生物制品檢定所提供,批號:121015-201004);川西獐牙菜對照藥材(中國藥品生物制品檢定所提供,批號:121354-200401);大黃對照藥材(中國藥品生物制品檢定所提供,批號:120984-201202)。薄層層析用硅膠G(化學純,青島海洋化工有限公司);甲醇為色譜純(美國Fisher公司),其他試劑均為分析純。
1.2 儀器設備
Shimadzu-LC 2010C全自動液相色譜儀,Shimadzu CLASS-VP V6.14SP1數據工作站 (日本島津制造所);LibrorAEG-200電子天平(日本島津制造所);WHF-203B暗箱式三用紫外分析儀 (上海精科實業有限公司);Milliporesimplicity-185超純水器 (美國密理博公司);KQ-100DE型數控超聲波發生器(昆山市超聲儀器有限公司)。
2 薄層色譜鑒別
2.1 川西獐牙菜的鑒別
取本品內容物5 g,加乙醇30 mL,超聲處理20 min,濾過,濾液加水15 mL和鹽酸2 mL,加熱回流1 h,放冷,加石油醚(60~90℃)40 mL振搖提取,提取液蒸干,殘渣加乙醇2 mL使溶解,作為供試品溶液。另取川西獐牙菜對照藥材2 g,加乙醇20 mL,加熱回流1 h,濾過,同法制成對照藥材溶液。再取齊墩果酸對照品,加乙醇制成每毫升含1 mg的溶液,作為對照品溶液。參照薄層色譜法(《中國藥典》2010年版一部附錄ⅥB)試驗,點樣量均為10 μL,采用硅膠G薄層板,展開劑為環己烷-丙酮-乙酸乙酯(5∶2∶1),展開,取出,晾干,顯色劑為10%的硫酸乙醇溶液,105℃加熱顯色。在與對照藥材和對照品色譜相應的位置上,供試品色譜顯相同顏色的斑點。
2.2 大黃的鑒別
取本品內容物2 g,加20 mL甲醇,超聲處理15 min,濾過,濾液揮干,用20 mL水,溶解后加鹽酸2 mL,加熱回流20 min,立即冷卻,用乙醚振搖提(2次 ×20 mL),合并乙醚液,蒸干,用1 mL三氯甲烷溶解后即為供試品溶液。取大黃對照藥材0.5 g,按供試品溶液制備方法處理成對照藥材溶液。再取大黃酸對照品,加甲醇制成每毫升含1 mg的溶液,作為對照品溶液。參照薄層色譜法(《中國藥典》2010年版一部附錄ⅥB)試驗,點樣量均為5 μL,采用硅膠G薄層板,展開劑為石油醚(30~60℃)-甲酸乙酯-甲酸(15∶5∶1)的上層溶液,展開,取出,晾干,置365 nm紫外光燈下檢視。供試品色譜中,在與對照藥材和對照品色譜相應的位置上,顯相同的橙黃色熒光斑點;置氨蒸氣中熏后,斑點變為紅色。
2.3 黃芪的鑒別
取本品內容物6 g,加甲醇80 mL,加熱回流1 h,放冷,濾過,濾液蒸干,殘渣加水50 mL使溶解,濾過,濾液用水飽和的正丁醇振搖提取 (3次×40 mL),合并正丁醇液,用氨試液洗滌2次,每次50 mL,取正丁醇液,再用正丁醇飽和的水洗滌(2次×50 mL),正丁醇液蒸干,殘渣加水5 mL使溶解,放冷,通過D101型大孔吸附樹脂(內徑1.5 cm,長12 cm),以水80 mL洗脫,棄去水液,再用40%乙醇50 mL洗脫,棄去洗脫液,繼用70%乙醇100 mL洗脫,收集洗脫液,蒸干,殘渣加甲醇1 mL使溶解,作為供試品溶液。另取黃芪甲苷對參照品,加甲醇制成每毫升含1 mg的溶液,作為對照品溶液。照薄層色譜法(《中國藥典》2010年版一部附錄ⅥB)試驗,點樣量均為10 μL,采用硅膠G薄層板,展開劑為三氯甲烷-乙酸乙酯-甲醇-水(15∶40∶22∶10)10℃以下放置的下層溶液,顯色劑為10%硫酸乙醇溶液,105℃加熱顯色。供試品色譜中,在與對照品色譜相應的位置上,顯相同顏色的斑點。
2.4 木香的鑒別
取本品內容物3 g,加乙醚15 mL,振搖5 min,放置2 h,濾過,濾液作為供試品溶液。另取木香對照藥材1 g,加乙醇10 mL,同法制成對照藥材溶液。參照薄層色譜法(《中國藥典》2010年版一部附錄ⅥB)試驗,點樣量均為10 μL,采用硅膠G 薄層板,展開劑為環己烷-丙酮(10∶3),展開,取出,晾干,顯色劑為5%香草醛硫酸溶液,105℃加熱顯。供試品色譜中,在與對照藥材色譜相應的位置上,顯相同顏色的斑點。
2.5 訶子的鑒別
取本品內容物2 g,加乙醇10 mL,超聲處理10 min,放置30 min,濾過,濾液作為供試品溶液。另取訶子對照藥材1 g,加乙醇5 mL,同供試品溶液制備方法制成對照藥材溶液。再取沒食子酸對照品,加乙醇制成每毫升含1 mg的溶液,作為對照品溶液。參照薄層色譜法(《中國藥典》2010年版一部附錄Ⅵ B)試驗,點樣量均為5 μL,采用硅膠 GF254 薄層板,展開劑為三氯甲烷-乙酸乙酯-甲酸(5∶4∶1),展開,取出,晾干,置254 nm紫外光燈下檢視。供試品色譜中,在與對照藥材和對照品色譜相應的位置上,顯相同顏色的熒光斑點。
3 大黃素和大黃酚含量測定[2]
3.1 色譜條件
Kromasil ODS-l色譜柱 (250 mm× 4.6 mm,5 μm);甲醇-0.1%磷酸溶液(85∶15)為流動相;柱溫:30℃;以 254 nm為檢測波長。
3.2 對照品溶液的制備
精密稱取大黃素、大黃酚對照品各5 mg,分別置50 mL量瓶中,用甲醇溶解并稀釋至刻度,搖勻;分別精密吸取大黃素溶液1 mL、大黃酚溶液2 mL,分別置50 mL量瓶中,加甲醇至刻度,搖勻,即得(大黃素每1 mL中含2 μg,大黃酚每毫升中含 4 μg)。
3.3 供試品溶液的制備
取本品約1.0 g,精密稱定,置100 mL錐形瓶中,加8%鹽酸20 mL,超聲處理5 min,再加三氯甲烷20 mL,加熱回流1 h,冷卻,移置分液漏斗中,用少量三氯甲烷洗滌容器,并入分液漏斗中,分取三氯甲烷層,酸液用三氯甲烷提取(2次×10 mL),合并三氯甲烷液,置水浴上揮干溶劑,殘渣加適量甲醇微熱使溶解,移入50 mL量瓶中,用少量甲醇分次洗滌容器,洗液移入同一量瓶中,加甲醇至刻度,搖勻,用微孔濾膜(0.45 μm)濾過,取續濾液,即得。
3.4 陰性對照溶液的制備
按本品處方比例稱取缺大黃的其余藥味,按生產工藝制成陰性對照,取陰性對照細粉約1 g,精密稱定,置100 mL具塞錐形瓶中,依供試品溶液制備方法項下自“加8%鹽酸20 mL……”起,制備陰性對照溶液。
在上述條件下,各蒽醌類成分與供試品溶液中其他色譜峰達到了基線分離,陰性對照試驗結果表明無干擾,色譜圖見圖1。
3.5 線性關系考察
3.5.1 大黃素線性范圍考察 精密稱取大黃素對照品適量,加甲醇制成每毫升含0.096 mg的溶液,搖勻,精密量取1 mL置25 mL量瓶中,加甲醇稀釋至刻度,搖勻,作為貯備液。分別精密量取貯備液1、2、3、4 mL置5 mL量瓶中,加甲醇稀釋至刻度,搖勻,分別制成每毫升含0.768、1.536、2.304、3.072 μg的溶液。分別精密吸取上述梯度濃度的對照品溶液與貯備液10 μL,注入液相色譜儀,記錄峰面積。以進樣濃度C(μg/mL)為橫坐標,峰面積A為縱坐標進行線性回歸,得標準曲線方程為:A=33319 C+1536.1,r=0.9996。表明在0.768~3.840 μg/mL范圍內,大黃素峰面積與進樣濃度具有良好的線性關系。
3.5.2 大黃酚線性范圍考察 精密稱取大黃酚對照品適量,加甲醇制成每毫升含0.102 mg的溶液,作為貯備液。分別精密量取貯備液 0.5、1、2、3、4 mL 置 25 mL量瓶中, 加甲醇分別制成每毫升含 2.04、4.08、8.16、12.24、16.32 μg 的溶液。分別精密吸取上述溶液各10 μL,注入液相色譜儀,記錄峰面積。以進樣濃度C(μg/mL)為橫坐標,峰面積A為縱坐標進行線性回歸,得標準曲線方程為:A=39750 C+22239,r= 0.9993。表明在 2.04~16.32 μg/mL 范圍內,大黃酚峰面積與進樣濃度具有良好的線性關系。
3.6 穩定性實驗
同一批樣品(批號:100402)按照上述供試品制備方法制成供試品溶液,在上述色譜條件下,每隔2 h進樣1次,每次10 μL,計算大黃素和大黃酚總量的RSD為0.49%。
3.7 精密度
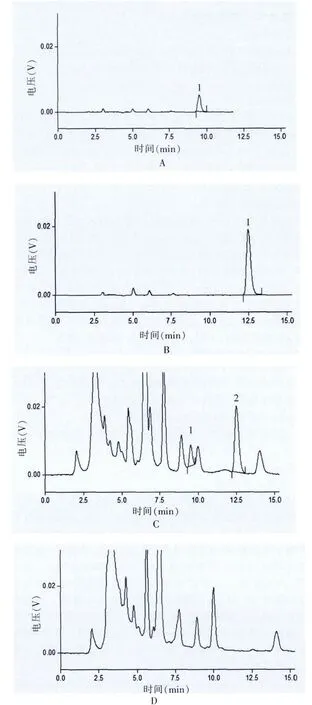
圖1 復方刺山柑膠囊中大黃HPLC圖譜
3.7.1 重復性 取同一份供試品溶液,在上述色譜條件下重復進樣5次,峰面積的RSD:大黃素為1.56%,大黃酚為1.24%。
3.7.2 日間精密度 取同一批樣品(批號:100402)分別在不同天(連續5 d),按照上述供試品制備方法制成供試品溶液,在上述色譜條件下測定,計算大黃素和大黃酚總量的RSD為0.70%。
3.8 回收率測定
3.8.1 大黃素 精密稱取供試品細粉0.5 g,共10份,其中一組為空白組,其余分三組,分別加入0.096 mg/mL的大黃素對照品溶液0.31、0.62、1.05 mL,照供試品溶液制備方法處理后,在上述色譜條件下進樣10 μL,計算回收量和回收率,結果表明,9次測定的平均回收率為100.00%(n=9),RSD 為 1.18%(n=9)。見表 1。
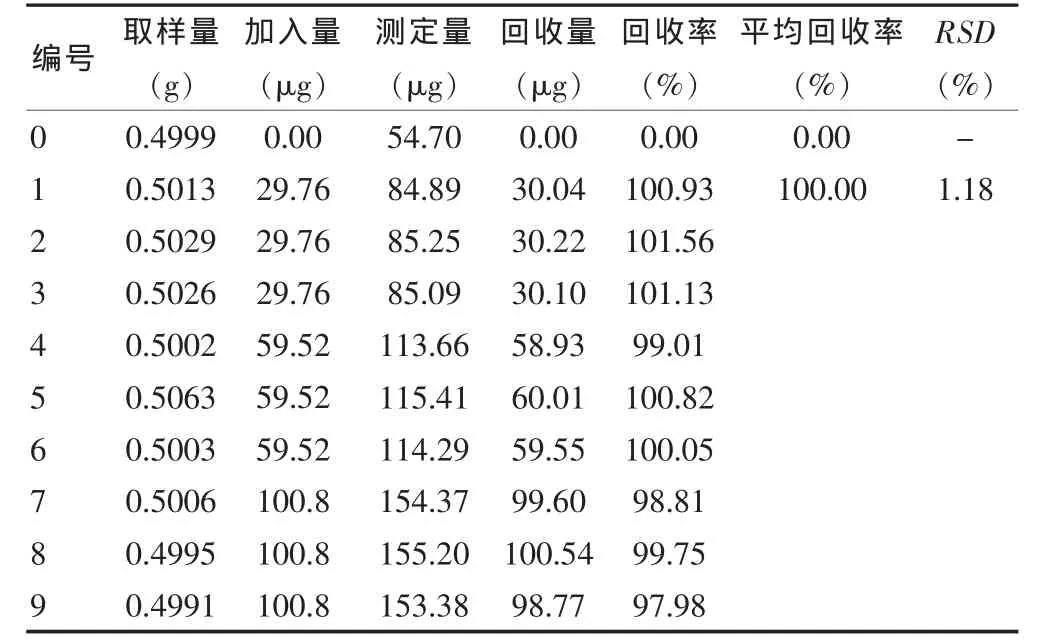
表1 大黃素回收率測定結果
3.8.2 大黃酚 精密稱取供試品細粉0.5 g,共10份,其中一組為空白組,其余分三組,分別加入0.102 mg/mL的大黃酚對照品溶液1、2、3.5 mL,照供試品溶液制備方法處理后,在上述色譜條件下進樣10 μL,計算回收量和回收率,9次測定的平均回收率為100.62%(n=9),RSD 為 0.38%(n=9)。見表2。
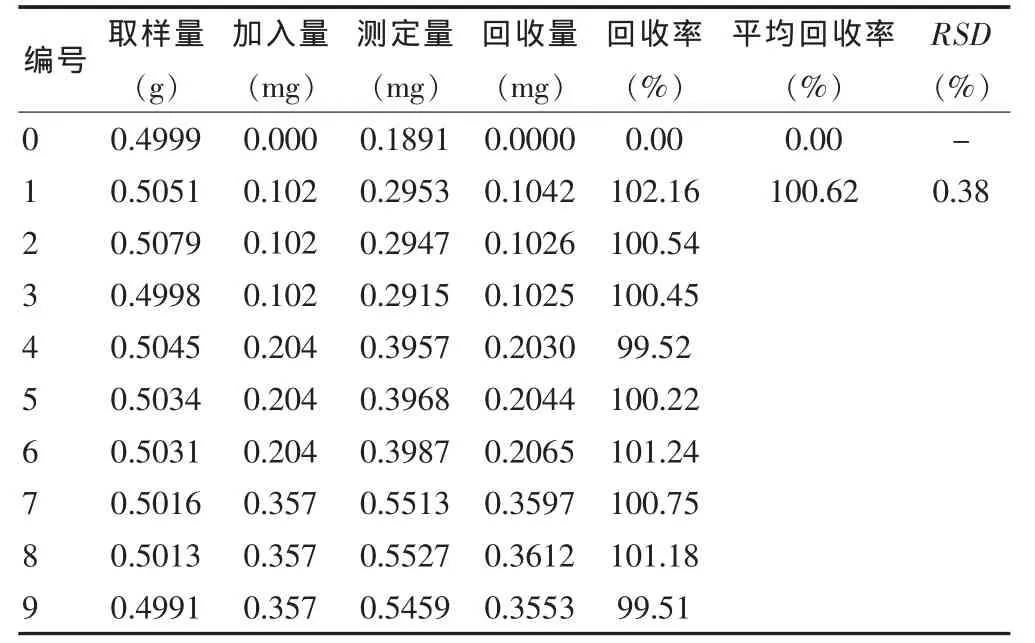
表2 大黃酚回收率測定結果
3.9 10批樣品測定
對制備的10批樣品(其中3批為中試規模)進行含量測定,每批樣品重復測定2次,計算平均值,結果見表3。
考慮到藥材的產地來源,以及制劑生產、儲藏等因素,暫定本品每克含大黃以大黃素和大黃酚總量計,不得低于0.35 mg。按每粒裝0.36 g計算,限度0.13 mg/粒。
4 討論
4.1 質量控制指標的選擇依據
選擇合理的質量控制指標是中藥復方藥物研究的關鍵點,也是難點,而將工藝篩選研究中的考察指標納入質量控制體系不僅能夠追蹤藥效成分或化學成分的轉移過程,而且有利于驗證工藝路線的穩定性和可行性。本品工藝研究中,有學者采用80%乙醇提取了川西獐芽菜、大黃、木香等藥味,其總蒽醌提取率達13.09~17.62 mg/g,正丁醇萃取物得率為13.31~16.32 mg/g,三氯甲烷萃取物得率達7.22~9.64 mg/g[1]。本文通過對大黃中兩種蒽醌類成分的測定,計算其轉移率達65%以上,與工藝研究結果吻合。川西獐芽菜[3-5]、大黃[6]、木香[7]中脂溶性成分具有保肝利膽作用,通過定性研究,對脂溶性有效成分進行了有效控制。針對訶子[8]、黃芪[9-10]中含大量多糖和鞣質類成分及與本方功能主治緊密結合的特點,提取工藝采用復方共煎,本文對二味進行TLC鑒別研究。本研究兼顧了提取工藝的考察指標,最大限度地保證了臨床的安全有效和質量可控。

表3 10批樣品中大黃素和大黃酚總量測定結果(mg/g)
4.2 最小檢測限、定量限測定
根據復方中大黃素和大黃酚含量較低的特點,研究中分別配制濃度適當的大黃素和大黃酚對照品溶液,在含量測定色譜條件下進樣10 μL,結果表明大黃素的最小檢測濃度約 0.261 μg/mL(信噪比 S/N=3),定量濃度約 0.384 μg/mL(信噪比S/N=10);大黃酚的最小檢測濃度約0.163 μg/mL(信噪比 S/N=3),定量濃度約 0.408 μg/mL(信噪比 S/N=10)。檢測結果表明本品測定濃度遠高于兩種含量測定指標的定量限,為定量測定提供了依據。
中藥復方的成分復雜,采用幾個指標成分或藥效成分進行定性定量研究難以全面控制產品質量,課題組對處方中刺山柑的化學成分和質量標準進行了研究[11-12],同時對其他藥味均進行了質量控制研究,因專屬性不強或無對照物質,無法作為質量控制指標。今后還需要深入研究其藥效物質基礎,建立能夠全面控制本品質量的標準。
[1]焦勝春,楊飛,楊偉俊,等.復方刺山柑膠囊提取工藝的優化[J].中成藥,2011,33(12):2169-2171.
[2]國家藥典委員會.中國藥典[S].一部.北京:中國醫藥科技出版社,2010:22-23.
[3]尚軍,張國燕,楊淳彬,等.川西獐牙菜的化學成分研究[J].青海師范大學學報自然科學版,2008,(4):66-67.
[4]羅桂花,趙建平,陳海娟,等.川西獐牙菜醇提物對小鼠CCl4肝損傷的保護作用[J].四川中醫,2008,26(11):29-30.
[5]呂坪,杜玉枝,李岑,等.川西獐牙菜醇提水沉部位抗黃疸性肝損傷的活性研究[J].時珍國醫國藥,2011,22(5):1098-1099.
[6]王耀先,郝慧南,陳秀瑋,等.大黃素通過內源性線粒體途徑和外源性死亡受體途徑誘導人宮頸癌Hela細胞凋亡的研究[J].中國中醫藥科技,2013,(1):37-39.
[7]許麗佳,章津銘,瞿燕,等.川木香醇提物利膽鎮痛作用的實驗研究[J].江蘇中醫藥,2010,42(9):76-77.
[8]Harpreet W,Rajbir S,Subodh K,et al.Effect of fractionation on antiradical efficacy of ethyl acetate extract of Terminalia chebula Retz[J].African Journal of Pharmacy and Pharmacology,2010,4(5):276-285.
[9]吳強,楊雁,薛紹禮,等.黃芪總苷對肝星狀細胞增殖和合成膠原的抑制作用[J].中國藥理學通報,2003,19(8):892.
[10]馬瑩,李潤琴,賈建偉,等.注射用黃芪多糖聯合肝動脈栓塞化療治療原發性肝癌療效觀察[J].中草藥,2008,39(12):1856-1858.
[11]趙軍,楊偉俊,任遠,等.刺山柑化學成分研究[J].天然產物研究與開發,2012,24(1):52-54,24.
[12]楊偉俊,阿不都沙拉木,陳燕,等.刺山柑果質量標準研究[J].時珍國醫國藥,2011,22(1):133-134.
